|
|
| |
proprio dello stato di aggregazione della materia nel quale gli atomi o le molecole sono distribuiti in maniera ordinata e tali da occupare nello spazio posizioni che si ripetono regolarmente formando il reticolo cristallino.
Comunemente lo stato s. viene definito anche come quello in cui i corpi presentano forma e volume propri. Molte sostanze, come i metalli, oltre ad avere una forma e un volume proprio, presentano anche una disposizione ordinata degli atomi (s. cristallini); altre invece, come i vetri, hanno una distribuzione casuale degli atomi nello spazio e pe 616f52g rciò sono detti s. amorfi. Dal punto di vista strutturale questi ultimi sono molto simili ai liquidi e possono essere considerati liquidi con elevato indice di viscosità o di attrito interno. Un'importante caratteristica dei s. è la rigidità e la conseguente reattività alle sollecitazioni esterne, come trazione, compressione, flessione, ecc., le quali inducono deformazioni che comportano variazioni di forma e di volume non sempre trascurabili.
Ruolo sempre più importante sta assumendo la fisica dello stato s., che studia le proprietà dei s. in relazione alla loro struttura intima sia dal punto di vista cristallografico (tipo di cella elementare, parametri reticolari, posizione nella cella degli atomi di specie diverse), sia per quanto concerne le forze che legano fra loro gli atomi nel reticolo cristallino. Essa cerca di correlare ai tipi di legami e alle loro intensità le specifiche proprietà dei s. come quelle di resistenza alle sollecitazioni meccaniche, di deformabilità, di conducibilità elettrica e termica, e altre. La fisica dei s., pur avendo avuto origine nei secoli passati, ha avuto una sua caratteristica linea di sviluppo dopo il 1928 con l'applicazione delle leggi della meccanica quantistica e statistica allo studio dei s., cioè quando, con la determinazione di basi teoriche definitive, si iniziò un processo di unificazione concettuale delle proprietà meccaniche, elettriche, magnetiche, dielettriche, ottiche, termiche dei solidi. I confini della fisica dei s. con altre branche della scienza come la metallurgia, l'elettronica, la chimica, ecc. che si occupano di materiali s. non sono definibili con precisione. La necessità di trovare soluzione a problemi di tipo tecnologico ha sempre rappresentato una grossa motivazione di tipo pratico per sviluppare ricerche nel campo della fisica dei solidi. Lo studio della diffrazione dei raggi X attraverso i s. ha messo in evidenza che la maggior parte di essi presenta struttura cristallina; sono cioè costituiti da atomi ordinati nello spazio secondo un reticolo. I reticoli cristallini possono presentare simmetrie svariate: cubica, tetragonale, ecc. Allo stato cristallino si contrappongono, in percentuale non rilevante, diversi stati amorfi, nei quali la materia non presenta le intime regolarità di struttura proprie della struttura cristallina. L'esistenza di una simmetria spaziale degli atomi è fondamentale per una trattazione teorica rigorosa dello stato s., da cui si deducono schemi che permettono l'interpretazione di proprietà fondamentali (come la teoria delle bande per la distinzione tra conduttori e isolanti). Nei cristalli a volte esistono imperfezioni o irregolarità nel reticolo le quali hanno grande importanza perché a esse sono legate particolari proprietà, come quelle dei semiconduttori, e caratteristici fenomeni ottici (luminescenza, effetto laser), plastici, di accrescimento dei cristalli, di diffusione, ecc. Lo studio dei cristalli ideali, cioè quelli immaginati senza imperfezioni, si occupa delle vibrazioni reticolari, del moto degli elettroni, delle interazioni con la radiazione, del ferromagnetismo, della superconduttività e definisce le cosiddette proprietà intrinseche del s. dipendenti esclusivamente dalla sua natura chimica e dalla sua struttura cristallina ideale. Le scoperte e gli studi sui semiconduttori, che hanno rivoluzionato l'elettronica, consentendo la miniaturizzazione dei circuiti, rappresentano uno dei capitoli più noti della fisica dei s., ma ormai l'approfondimento e l'estensione della materia sono tali che influenzano e sono influenzati da sempre più numerosi campi di ricerca (fisica nucleare, energia nucleare, scienze spaziali, elettronica, ecc.) che richiedono particolari sviluppi della tecnologia dei materiali.
DETERMINAZIONE DEL COMPORTAMENTO DINAMICO DEGLI ELETTRONI LIBERI NEL RETICOLO CRISTALLINO
Quantisticamente il comportamento dinamico degli elettroni liberi nel reticolo cristallino viene determinato da una funzione d'onda che soddisfa l'equazione di Schrödinger che, date le caratteristiche del potenziale creato dagli atomi del reticolo, è assai complessa. In generale, pertanto, si introducono modelli semplificati del potenziale. Se si considera un reticolo cristallino unidimensionale, la soluzione dell'equazione di Schrödinger mostra che gli elettroni possono avere solo energie di valore compreso entro intervalli ben definiti, chiamati bande permesse, separati da bande di energia proibite, dette appunto bande proibite. In una rappresentazione in cui si esprima l'energia degli elettroni in funzione del numero d'onda, cioè dell'inverso della lunghezza d'onda associata all'elettrone, queste bande sono dette zone di Brillouin. Risultati analoghi si ottengono per un reticolo cristallino tridimensionale se si considera il numero d'onda come una grandezza vettoriale. All'interno di ciascuna banda gli elettroni possono assumere solo un certo numero di valori discreti dell'energia, ciascuno dei quali costituisce un livello energetico. Data una banda con N livelli, questa, per il principio di esclusione di Pauli, può contenere 2N elettroni. Nelle bande complete, cioè contenenti 2N elettroni, la velocità media di questi ultimi è nulla e questi non contribuiscono pertanto alla corrente che circola nel corpo; nelle bande occupate solo parzialmente l'assenza di un elettrone equivale alla presenza di una carica identica, ma di segno opposto, cioè positiva, chiamata buco. La distribuzione degli elettroni in una banda è regolata dalla statistica di Fermi-Dirac e in essa è fondamentale il livello di Fermi che corrisponde al massimo valore dell'energia che può venire raggiunta dagli elettroni alla temperatura di.0 K.
In questo stato di aggregazione le molecole presentano forze di coesione che tendono a mantenerle aderenti tra loro senza però impedire lo scorrimento delle une sulle altre. I l. non hanno quindi una forma propria ma assumono quella del recipiente che li contiene.
Lo stato l. può considerarsi come uno stato intermedio tra quello solido e quello aeriforme e può sempre verificarsi il passaggio dall'uno all'altro dei tre stadi. In un l. in riposo il moto di scorrimento delle molecole le une sulle altre è continuo e si identifica con il moto di agitazione termica. In tale moto vengono a formarsi tra le molecole, per dei tempi brevissimi, cavità con diametri dell'ordine delle dimensioni molecolari che subito si richiudono riformandosi altrove; di conseguenza, i l. presentano in genere una densità di poco inferiore a quella del solido nel quale ciascuno di essi si trasforma per raffreddamento e sono assai poco comprimibili. Le forze di coesione che agiscono tra le molecole dei l. sono di diversa intensità e variano con la temperatura, dando corrispondentemente luogo a valori della viscosità diversi secondo il l. considerato e secondo la temperatura. Tali forze possono, inoltre, essere di diversa natura: di natura prevalentemente elettrostatica come nei sali fusi, in pratica costituiti da ioni di carica opposta, oppure del tipo delle forze cosiddette di van der Waals che si esercitano tra molecole elettricamente neutre o ancora (p. es. nel caso dell'acqua) dovute alla struttura polare delle molecole del l. che comporta l'esistenza, all'interno di ogni molecola, dizone a prevalente carica positiva accanto ad altre a prevalente carica negativa. Una molecola di un l. che in un dato istante non sia immediatamente adiacente alla superficie che separa questo dall'atmosfera soprastante si trova soggetta alle forze attrattive esercitate dalle altre molecole che la circondano in tutte le direzioni; le molecole che in un dato istante costituiscono la superficie del l. sono invece adiacenti a un numero di molecole minori e quelle di esse che in quell'istante presentano un'energia cinetica più elevata possono passare dalla massa liquida all'atmosfera soprastante, trasformandosi cioè in vapore. Ne deriva che tutti i l., anche a temperature largamente inferiori al loro punto di ebollizione, presentano una tendenza più o meno spiccata a evaporare: tale tendenza aumenta con la temperatura perché con questa aumenta l'energia cinetica delle molecole. Dal vapore che viene così a formarsi al disopra della superficie liquida, se questo non viene di continuo asportato (p. es. ventilando la superficie stessa), le molecole presentano una certa tendenza a ritornare nella massa liquida, verso la quale la richiamano le forze attrattive prima descritte. A ogni temperatura viene così a stabilirsi un equilibrio nel quale si uguagliano il numero di molecole di l. che, in un certo istante, evaporano e il numero di molecole che contemporaneamente condensano, ossia ritornano dallo stato gassoso a quello liquido. La pressione del vapore del l., in queste condizioni, prende il nome di tensione di vapore del l. stesso e aumenta con la temperatura in ragione logaritmica.
Sostanza liquida contenente in sospensione particelle di materiale magnetico di dimensioni nell'ordine del millesimo di millimetro; a volte viene chiamato anche ferrofluido. Caratteristica di questi l. è l'elevatissima sensibilità ai campi magnetici, questo perché ciascuna particella in sospensione nel l. possiede un momento magnetico proprio, cioè si comporta come una sorta di micromagnete. I principali interessi sui l. magnetici risiedono nelle loro proprietà ottiche: p. es. poiché le particelle di un l. magnetico si orientano in presenza di un campo magnetico è possibile utilizzare campi magnetici per polarizzare opportunamente questi l. e far sì, di volta in volta, che siano trasparenti o opachi alla luce. Questo permette di misurare la viscosità dei l. (fig. 1) e la presenza di deboli campi magnetici (fig. 2). In medicina sono utilizzati nella localizzazione di cellule cancerose e nell'industria elettronica dove vengono usati per la realizzazione di stampanti ad alta velocità.
Il gas è aeriforme non condensabile a temperatura ambiente, cioè avente temperatura critica decisamente più bassa di quelle normalmente riscontrabili negli ambienti naturali. Il termine è usato talvolta, ma improprio, per indicare gli aeriformi; stato gassoso è anche detto lo stato di aggregazione della materia che corrisponde allo stato aeriforme. Lo studio dei g. è molto facilitato dalla considerazione di un modello ideale detto g. perfetto, oggetto sia della termodinamica, sia della teoria cinetica dei g. e della meccanica statistica.
Lo studio dei g. si è
storicamente sviluppato nell'ambito della termodinamica la quale considera
variabili quali pressione, volume e temperatura che prescindono dalla
conoscenza della costituzione microscopica dei g. e addirittura dal fatto che
questi ultimi siano formati da atomi o da molecole. Appunto nell'ambito della
termodinamica, che considera solo grandezze macroscopiche, si deducono le leggi
di Boyle e Mariotte, di Charles e Gay-Lussac e l'equazione di stato dei g.
perfetti, nonché il valore numerico della costante universale dei g., R.
Un g. perfetto viene definito dalla teoria cinetica dei g., come un aeriforme
ideale il cui comportamento sia perfettamente aderente a quello previsto da
queste leggi. La legge di Boyle-Mariotte rappresenta le trasformazioni
isotermiche (a temperatura costante) del g. perfetto; quelle di Charles e
Gay-Lussac rappresentano le trasformazioni isobariche (a pressione costante) e
isocore (a volume costante). La considerazione contemporanea di più di una di
queste trasformazioni porta alla legge delle trasformazioni adiabatiche (senza
scambio di calore): pVg=costante
e all'equazione caratteri stica dei g. perfetti: ![]() _[1v\h8"f10_83.wmf"_[0v. In
queste relazioni p, V e T indicano rispettivamente
pressione, volume e temperatura assoluta; g indica il rapporto tra calore specifico del g. a pressione
costante e calore specifico a volume costante; p e V
indicano rispettivamente la pressione di una atmosfera e il volume occupato da
una grammomolecola di g. a quella pressione e alla temperatura di 0 ºC, cioè
22,414 litri; n è il numero di grammomolecole considerate e 273,15 il
coefficiente di dilatazione termica dei gas.
_[1v\h8"f10_83.wmf"_[0v. In
queste relazioni p, V e T indicano rispettivamente
pressione, volume e temperatura assoluta; g indica il rapporto tra calore specifico del g. a pressione
costante e calore specifico a volume costante; p e V
indicano rispettivamente la pressione di una atmosfera e il volume occupato da
una grammomolecola di g. a quella pressione e alla temperatura di 0 ºC, cioè
22,414 litri; n è il numero di grammomolecole considerate e 273,15 il
coefficiente di dilatazione termica dei gas. ![]() _[1v\h8"f10_84.wmf"_[0v è
pertanto una costante, detta costante universale dei g. perfetti, che vale, nel
Sistema Internazionale (S.I.), 8,31 joule/kelvin mole. L'equazione caratteristica dei g. perfetti si scrive,
pertanto, pV=nRT. Lo studio dei g., in via puramente teorica, potrebbe
essere effettuato applicando alle singole molecole costituenti le leggi della
meccanica classica; per questa via si dovrebbe pervenire alle stesse leggi
dedotte per via puramente termodinamica. In pratica, dato il numero
estremamente elevato delle molecole di un g., che occupano i volumi
considerati, lo studio di un sistema così complesso non può essere effettuato
neppure con l'aiuto di un elaboratore elettronico comunque potente. Si trova
però che per la deduzione delle leggi dei g., usando le equazioni della
dinamica, non è necessario conoscere il comportamento di ogni singola molecola,
ma è solo necessario dedurre il valore medio delle grandezze fisiche relative
alle molecole costituenti: si possono cioè considerare tutte le variabili
termodinamiche come medie effettuate sulle grandezze meccaniche microscopiche.
Le leggi della meccanica vengono in effetti applicate statisticamente a sistemi
complessi quali i g., a due diversi livelli di complessità formale. Il primo, e
il più complesso, sviluppato particolarmente da J. W. Gibbs e da L. Boltzmann,
utilizza tecniche matematiche molto astratte e, in linea di principio, staccate
dalla realtà fisica a cui si riferiscono: è la meccanica statistica che trova
applicazione anche nel caso di sistemi per i quali non sono più applicabili le
leggi della meccanica classica; in questo caso si parla generalmente di
meccanica statistica quantistica e i sistemi quantizzati possono essere anche i
g. di elettroni in un atomo o in un metallo. Il secondo, sviluppato soprattutto
da R. Boyle, D. Bernoulli, J. Joule, R. Clausius e C. Maxwell, costituisce la teoria
cinetica dei gas. Essa si avvale particolarmente di considerazioni di
carattere fisico e si può considerare come una prima approssimazione della
stessa meccanica statistica, per quanto la sua caratteristica principale
consista nello stabilire relazioni e corrispondenze tra grandezze fisiche macroscopiche
(quelle della termodinamica) e grandezze fisiche microscopiche (quelle
meccaniche, relative alle singole molecole del gas). Le ipotesi fondamentali
della teoria cinetica sono le seguenti: un g. è costituito da particelle,
chiamate molecole, che si muovono di moto casuale in tutte le direzioni, con
diverse velocità, e per le quali sono perfettamente valide le leggi della
meccanica classica; in un volume, anche piccolissimo, di g. esiste sempre un
numero estremamente grande di molecole; il diametro delle molecole è
trascurabile rispetto alla distanza media tra esse; le traiettorie delle
molecole sono rettilinee e vengono modificate solo dagli urti; gli urti, che
sono perfettamente elastici, cioè avvengono senza perdita di energia cinetica,
hanno una durata assolutamente trascurabile rispetto al tempo durante il quale
le molecole si muovono liberamente; all'interno del g., la distribuzione delle
molecole costituenti è totalmente disordinata, per cui in volumi uguali, anche
piccolissimi, ma sempre contenenti un numero enorme di molecole, si trovano
numeri uguali di molecole. In base a queste ipotesi si può, p. es., calcolare
la pressione, p, esercitata da un g. sulle pareti del recipiente che lo
contiene:
_[1v\h8"f10_84.wmf"_[0v è
pertanto una costante, detta costante universale dei g. perfetti, che vale, nel
Sistema Internazionale (S.I.), 8,31 joule/kelvin mole. L'equazione caratteristica dei g. perfetti si scrive,
pertanto, pV=nRT. Lo studio dei g., in via puramente teorica, potrebbe
essere effettuato applicando alle singole molecole costituenti le leggi della
meccanica classica; per questa via si dovrebbe pervenire alle stesse leggi
dedotte per via puramente termodinamica. In pratica, dato il numero
estremamente elevato delle molecole di un g., che occupano i volumi
considerati, lo studio di un sistema così complesso non può essere effettuato
neppure con l'aiuto di un elaboratore elettronico comunque potente. Si trova
però che per la deduzione delle leggi dei g., usando le equazioni della
dinamica, non è necessario conoscere il comportamento di ogni singola molecola,
ma è solo necessario dedurre il valore medio delle grandezze fisiche relative
alle molecole costituenti: si possono cioè considerare tutte le variabili
termodinamiche come medie effettuate sulle grandezze meccaniche microscopiche.
Le leggi della meccanica vengono in effetti applicate statisticamente a sistemi
complessi quali i g., a due diversi livelli di complessità formale. Il primo, e
il più complesso, sviluppato particolarmente da J. W. Gibbs e da L. Boltzmann,
utilizza tecniche matematiche molto astratte e, in linea di principio, staccate
dalla realtà fisica a cui si riferiscono: è la meccanica statistica che trova
applicazione anche nel caso di sistemi per i quali non sono più applicabili le
leggi della meccanica classica; in questo caso si parla generalmente di
meccanica statistica quantistica e i sistemi quantizzati possono essere anche i
g. di elettroni in un atomo o in un metallo. Il secondo, sviluppato soprattutto
da R. Boyle, D. Bernoulli, J. Joule, R. Clausius e C. Maxwell, costituisce la teoria
cinetica dei gas. Essa si avvale particolarmente di considerazioni di
carattere fisico e si può considerare come una prima approssimazione della
stessa meccanica statistica, per quanto la sua caratteristica principale
consista nello stabilire relazioni e corrispondenze tra grandezze fisiche macroscopiche
(quelle della termodinamica) e grandezze fisiche microscopiche (quelle
meccaniche, relative alle singole molecole del gas). Le ipotesi fondamentali
della teoria cinetica sono le seguenti: un g. è costituito da particelle,
chiamate molecole, che si muovono di moto casuale in tutte le direzioni, con
diverse velocità, e per le quali sono perfettamente valide le leggi della
meccanica classica; in un volume, anche piccolissimo, di g. esiste sempre un
numero estremamente grande di molecole; il diametro delle molecole è
trascurabile rispetto alla distanza media tra esse; le traiettorie delle
molecole sono rettilinee e vengono modificate solo dagli urti; gli urti, che
sono perfettamente elastici, cioè avvengono senza perdita di energia cinetica,
hanno una durata assolutamente trascurabile rispetto al tempo durante il quale
le molecole si muovono liberamente; all'interno del g., la distribuzione delle
molecole costituenti è totalmente disordinata, per cui in volumi uguali, anche
piccolissimi, ma sempre contenenti un numero enorme di molecole, si trovano
numeri uguali di molecole. In base a queste ipotesi si può, p. es., calcolare
la pressione, p, esercitata da un g. sulle pareti del recipiente che lo
contiene: ![]() _[1v\h8"f10_85.wmf"_[0v in cui m
è la massa di una molecola, n è il numero di molecole per unità di
volume (e pertanto mn è la densità del g.) e v è la velocità quadratica media delle molecole definita
come
_[1v\h8"f10_85.wmf"_[0v in cui m
è la massa di una molecola, n è il numero di molecole per unità di
volume (e pertanto mn è la densità del g.) e v è la velocità quadratica media delle molecole definita
come
![]() _[1v\h8"f10_86.wmf"_[0v
_[1v\h8"f10_86.wmf"_[0v
essendo N il numero complessivo di
molecole e vxi, vyi, vzi le
componenti delle velocità dell'i-esima molecola secondo gli assi di una
terna cartesiana. Si noti come, in questa relazione, il valore medio di una
grandezza microscopica, la velocità delle molecole, è legato a grandezze
macroscopiche misurabili, densità e pressione del gas. Analogamente, si
perviene a una relazione tra velocità quadratica media e temperatura assoluta, ![]() _[1v\h8"f10_87.wmf"_[0v, in cui
M è il peso molecolare del g. e R è la costante universale dei
gas. Questa relazione permette anche di affermare che l'energia cinetica totale
delle molecole del g. è proporzionale alla sua temperatura assoluta. Inoltre,
dividendo ambo i membri della relazione precedente per il numero di Avogadro si
trova:
_[1v\h8"f10_87.wmf"_[0v, in cui
M è il peso molecolare del g. e R è la costante universale dei
gas. Questa relazione permette anche di affermare che l'energia cinetica totale
delle molecole del g. è proporzionale alla sua temperatura assoluta. Inoltre,
dividendo ambo i membri della relazione precedente per il numero di Avogadro si
trova: ![]() _[1v\h8"f10_88.wmf"_[0v in cui k
è la costante di Boltzmann e m la massa di una singola molecola. Un
altro importante risultato della teoria cinetica si ha nella considerazione dei
calori specifici di un g. a pressione costante, cp, e a volume costante, cv. Nei g., il calore specifico a pressione costante è sempre
maggiore del calore specifico a volume costante; infatti, se si cede calore a
un g. mantenendone costante la pressione, il calore ceduto viene solo in parte
utilizzato per aumentare la temperatura del g., mentre la parte restante viene
spesa in lavoro meccanico fatto dal g. per espandersi contro le forze esterne.
Se, invece, al g. viene ceduto calore mantenendone costante il volume, il
calore viene tutto utilizzato per aumentarne la temperatura. Pertanto, nel
riscaldamento a pressione costante, è necessaria una quantità di calore
maggiore che a volume costante per aumentare di un grado la temperatura del g.
in esame: cp è quindi maggiore di cv. Se le molecole sono paragonabili a sferette, l'energia
cinetica del g. è tutta di traslazione, per cui l'energia interna U del
g. coincide con la somma delle energie cinetiche delle N molecole che lo
compongono:
_[1v\h8"f10_88.wmf"_[0v in cui k
è la costante di Boltzmann e m la massa di una singola molecola. Un
altro importante risultato della teoria cinetica si ha nella considerazione dei
calori specifici di un g. a pressione costante, cp, e a volume costante, cv. Nei g., il calore specifico a pressione costante è sempre
maggiore del calore specifico a volume costante; infatti, se si cede calore a
un g. mantenendone costante la pressione, il calore ceduto viene solo in parte
utilizzato per aumentare la temperatura del g., mentre la parte restante viene
spesa in lavoro meccanico fatto dal g. per espandersi contro le forze esterne.
Se, invece, al g. viene ceduto calore mantenendone costante il volume, il
calore viene tutto utilizzato per aumentarne la temperatura. Pertanto, nel
riscaldamento a pressione costante, è necessaria una quantità di calore
maggiore che a volume costante per aumentare di un grado la temperatura del g.
in esame: cp è quindi maggiore di cv. Se le molecole sono paragonabili a sferette, l'energia
cinetica del g. è tutta di traslazione, per cui l'energia interna U del
g. coincide con la somma delle energie cinetiche delle N molecole che lo
compongono: ![]() _[1v\h8"f10_89.wmf"_[0v. Da
questa re- lazione insieme alla cp=R+cv, dedotta per via puramente termodinamica e nella quale cp e cv sono
intesi come calo ri specifici molari, si ricava
_[1v\h8"f10_89.wmf"_[0v. Da
questa re- lazione insieme alla cp=R+cv, dedotta per via puramente termodinamica e nella quale cp e cv sono
intesi come calo ri specifici molari, si ricava ![]() _[1v\h8"f10_90.wmf"_[0v e
quindi:
_[1v\h8"f10_90.wmf"_[0v e
quindi: ![]() _[1v\h8"f10_90a.wmf"_[0v,
ovvero:
_[1v\h8"f10_90a.wmf"_[0v,
ovvero: ![]() _[1v\h8"f10_90b.wmf"_[0v. La
teoria cinetica permette quindi di prevedere il valore dei calori specifici
molari, cosa impossibile nell'ambito della sola termodinamica. I valori trovati
sperimentalmente sono però in buon accordo con quelli della teoria solo per g.
monoatomici, mentre per g. poliatomici è necessario introdurre un modello di
molecola non assimilabile a una sferetta. In una molecola biatomica, p. es., il
modello più aderente alla realtà è quello di un'asta con due sfere agli
estremi: in questo caso c'è un altro modo in cui la molecola può immagazzinare
energia cinetica. L'energia termica ceduta al g. può infatti trasformarsi,
oltre che in energia cinetica di traslazione, in energia cinetica di rotazione
attorno a un asse perpendicolare a quello congiungente i due atomi. L'energia
di rotazione attorno a quest'ultimo asse, come risulta da semplici
considerazioni di meccanica classica, è invece trascurabile. D'altra parte, si
può dimostrare con metodi statistici che l'energia disponibile per il g. si
distribuisce in parti uguali per ciascuno dei modi diversi in cui le molecole
possono assorbire energia; in altri termini, si distribuisce equamente tra i diversi
gradi di libertà del sistema (teorema di equipartizione dell'energia). Per via
termodinamica si ricava allora:
_[1v\h8"f10_90b.wmf"_[0v. La
teoria cinetica permette quindi di prevedere il valore dei calori specifici
molari, cosa impossibile nell'ambito della sola termodinamica. I valori trovati
sperimentalmente sono però in buon accordo con quelli della teoria solo per g.
monoatomici, mentre per g. poliatomici è necessario introdurre un modello di
molecola non assimilabile a una sferetta. In una molecola biatomica, p. es., il
modello più aderente alla realtà è quello di un'asta con due sfere agli
estremi: in questo caso c'è un altro modo in cui la molecola può immagazzinare
energia cinetica. L'energia termica ceduta al g. può infatti trasformarsi,
oltre che in energia cinetica di traslazione, in energia cinetica di rotazione
attorno a un asse perpendicolare a quello congiungente i due atomi. L'energia
di rotazione attorno a quest'ultimo asse, come risulta da semplici
considerazioni di meccanica classica, è invece trascurabile. D'altra parte, si
può dimostrare con metodi statistici che l'energia disponibile per il g. si
distribuisce in parti uguali per ciascuno dei modi diversi in cui le molecole
possono assorbire energia; in altri termini, si distribuisce equamente tra i diversi
gradi di libertà del sistema (teorema di equipartizione dell'energia). Per via
termodinamica si ricava allora: ![]() _[1v\h8"f10_91.wmf"_[0v, valore
di nuovo in buon ac- cordo con i valori ottenuti sperimentalmente per molecole
biatomiche. Un altro modo in cui le molecole biatomiche possono immagazzinare
energia è in moti di vibrazione lungo l'asse congiungente gli atomi. Un modello
di questo tipo è ancora soddisfacente e si trova no sperimentalmente valori di
_[1v\h8"f10_91.wmf"_[0v, valore
di nuovo in buon ac- cordo con i valori ottenuti sperimentalmente per molecole
biatomiche. Un altro modo in cui le molecole biatomiche possono immagazzinare
energia è in moti di vibrazione lungo l'asse congiungente gli atomi. Un modello
di questo tipo è ancora soddisfacente e si trova no sperimentalmente valori di ![]() _[1v\h8"f10_92.wmf"_[0vcorrispondenti
a quelli dedotti teoricamente. Ciò che è invece in sostanziale contrasto con la
teoria, e che può essere spiegato solo nell'ambito della meccanica quantistica,
è che il teorema di equipartizione dell'energia non è valido a tutte le
temperature, ma come si ricava sperimentalmente per una molecola biatomica come
l'idrogeno, il g. a basse temperature assorbe energia solo sotto forma di
energia cinetica traslazionale, a temperature maggiori sotto forma di energia
cinetica traslazionale e di energia cinetica rotazionale, a temperature ancora
maggiori sotto forma di energia cinetica traslazionale, di energia cinetica
rotazionale e di energia cinetica vibrazionale. L'energia dovrebbe invece,
secondo la teoria cinetica classica, distribuirsi sempre in egual misura nelle tre
diverse forme. Molte altre notevoli previsioni della teoria cinetica dei g.
hanno invece ottenuto piena conferma dai risultati sperimentali. A) Se si
considera la molecola come dotata di dimensioni finite, e non puntiforme, si
può dedurre la distanza percorsa in media dalla molecola di un g. nel tempo
intercorrente tra due urti successivi (cammino libero medio, l):
_[1v\h8"f10_92.wmf"_[0vcorrispondenti
a quelli dedotti teoricamente. Ciò che è invece in sostanziale contrasto con la
teoria, e che può essere spiegato solo nell'ambito della meccanica quantistica,
è che il teorema di equipartizione dell'energia non è valido a tutte le
temperature, ma come si ricava sperimentalmente per una molecola biatomica come
l'idrogeno, il g. a basse temperature assorbe energia solo sotto forma di
energia cinetica traslazionale, a temperature maggiori sotto forma di energia
cinetica traslazionale e di energia cinetica rotazionale, a temperature ancora
maggiori sotto forma di energia cinetica traslazionale, di energia cinetica
rotazionale e di energia cinetica vibrazionale. L'energia dovrebbe invece,
secondo la teoria cinetica classica, distribuirsi sempre in egual misura nelle tre
diverse forme. Molte altre notevoli previsioni della teoria cinetica dei g.
hanno invece ottenuto piena conferma dai risultati sperimentali. A) Se si
considera la molecola come dotata di dimensioni finite, e non puntiforme, si
può dedurre la distanza percorsa in media dalla molecola di un g. nel tempo
intercorrente tra due urti successivi (cammino libero medio, l): ![]() _[1v\h8"f10_93.wmf"_[0v, in cui
r è il raggio della molecola e n il numero di molecole per unità
di volume. B) Essendo estremamente grande il numero di molecole costituenti un
g., esiste una probabilità finita che ci sia un certo numero di molecole che
possiedono una velocità v data, qualsiasi. Tale numero dipende dalla
velocità prescelta e la distribuzione delle velocità, dedotta da C. Maxwell, è
data dall'espressione:
_[1v\h8"f10_93.wmf"_[0v, in cui
r è il raggio della molecola e n il numero di molecole per unità
di volume. B) Essendo estremamente grande il numero di molecole costituenti un
g., esiste una probabilità finita che ci sia un certo numero di molecole che
possiedono una velocità v data, qualsiasi. Tale numero dipende dalla
velocità prescelta e la distribuzione delle velocità, dedotta da C. Maxwell, è
data dall'espressione:
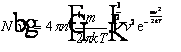 _[1v\h8"f10_94.wmf"_[0v
_[1v\h8"f10_94.wmf"_[0v
in cui N(v) è il numero di molecole aventi velocità v, n è il numero totale di molecole e m è la massa di ciascuna molecola. In figura sono riportate le distribuzioni di velocità per un g. costituito da 10 molecole di ossigeno nei due casi in cui la temperatura sia uguale a 73 K e a 273 K. C) Essendo i g. costituiti da molecole, una particella in sospensione in un g. deve partecipare ai moti di agitazione termica delle molecole ed essere dotata di un'energia cinetica media di traslazione uguale a (3/2) kT, in accordo con il teorema di equipartizione dell'energia. Tale previsione, fatta da A. Einstein prima che egli avesse conoscenza dell'esistenza dei moti browniani, fu confermata da misure molto precise sui moti browniani e da altre ancora più accurate effettuate studiando il moto casuale di leggerissimi specchi sospesi a fili di torsione e sottoposti agli urti delle particelle di g. dell'ambiente circostante. Fu questa la prima conferma dell'oggettiva esistenza delle molecole.
A differenza di quanto si suppone
per il g. perfetto, nei g. che effettivamente esistono cioè non ideali e che si
indicano con il nome di g. reali, le molecole presentano dimensioni definite,
anche se estremamente piccole, e inoltre si influenzano tra di loro con forze
di tipo sia attrattivo sia repulsivo, delle quali prevalgono, secondo i casi,
le une o le altre. Di conseguenza, il comportamento dei g. reali differisce in
maggior o minor misura da quello previsto dall'equazione di stato dei g.
perfetti. Le deviazioni dal comportamento ideale sono più spiccate per i g. di
peso molecolare relativamente elevato (nei quali ci si discosta maggiormente
dall'ipotesi che le molecole siano puntiformi) e nei g. a pressione molto elevata
(nei quali, essendo minore la distanza media tra le molecole, sono più intense
le forze che agiscono tra le molecole); in caso contrario le deviazioni sono
percentualmente ridotte, in genere assai inferiori all'1% e l'equazione di
stato del g. perfetto può venire applicata in prima approssimazione ai g.
reali. Per descrivere più fedelmente il comportamento di questi sono state
formulate varie equazioni di stato che introducono in quella del g. perfetto
delle correzioni connesse alla natura del gas. La più nota è quella di van der
Waals: ![]() _[1v\h8"f10_95.wmf"_[0v nella
quale il termine correttivo a/V , che si
indica con il nome di pressione interna, tiene conto delle forze
intermolecolari, mentre il termine b, che si indica con il nome di
covolume, tiene conto del volume proprio delle molecole, ossia del fatto che
esse non siano puntiformi. A temperatura ambiente tutti i g., eccetto
l'idrogeno, l'elio e il neo, fino a una pressione dell'ordine di qualche
centinaio di atmosfere, occupano un volume inferiore a quello calcolato per il
g. perfetto, ossia risultano più comprimibili di questo; l'idrogeno, l'elio e
il neo, alla stessa temperatura, qualunque sia la pressione, presentano invece
un volume maggiore di quello del g. perfetto, ossia risultano meno comprimibili.
In realtà, per ciascun g. reale esiste una temperatura, detta temperatura di
Boyle, alla quale il g. passa dall'uno all'altro comportamento: l'idrogeno,
l'elio e il neo differiscono quindi dagli altri g. solo per il fatto che la
loro temperatura di Boyle è molto inferiore alla temperatura ambiente. Un g.
reale che si trova a una temperatura superiore alla sua temperatura di Boyle e
a una pressione non eccessivamente elevata, quando si espande senza compiere
lavoro si raffredda: al di sopra della temperatura di Boyle tra le molecole del
g. si esercitano infatti soprattutto delle forze attrattive e per vincerle
viene assorbita una certa quantità di energia che si traduce appunto in un
abbassamento della temperatura del gas. Nel g. perfetto, nel quale per
definizione tra le molecole del g. non si manifestano forze né attrattive né
repulsive, un'espansione del genere non produrrebbe invece alcuna variazione di
temperatura.
_[1v\h8"f10_95.wmf"_[0v nella
quale il termine correttivo a/V , che si
indica con il nome di pressione interna, tiene conto delle forze
intermolecolari, mentre il termine b, che si indica con il nome di
covolume, tiene conto del volume proprio delle molecole, ossia del fatto che
esse non siano puntiformi. A temperatura ambiente tutti i g., eccetto
l'idrogeno, l'elio e il neo, fino a una pressione dell'ordine di qualche
centinaio di atmosfere, occupano un volume inferiore a quello calcolato per il
g. perfetto, ossia risultano più comprimibili di questo; l'idrogeno, l'elio e
il neo, alla stessa temperatura, qualunque sia la pressione, presentano invece
un volume maggiore di quello del g. perfetto, ossia risultano meno comprimibili.
In realtà, per ciascun g. reale esiste una temperatura, detta temperatura di
Boyle, alla quale il g. passa dall'uno all'altro comportamento: l'idrogeno,
l'elio e il neo differiscono quindi dagli altri g. solo per il fatto che la
loro temperatura di Boyle è molto inferiore alla temperatura ambiente. Un g.
reale che si trova a una temperatura superiore alla sua temperatura di Boyle e
a una pressione non eccessivamente elevata, quando si espande senza compiere
lavoro si raffredda: al di sopra della temperatura di Boyle tra le molecole del
g. si esercitano infatti soprattutto delle forze attrattive e per vincerle
viene assorbita una certa quantità di energia che si traduce appunto in un
abbassamento della temperatura del gas. Nel g. perfetto, nel quale per
definizione tra le molecole del g. non si manifestano forze né attrattive né
repulsive, un'espansione del genere non produrrebbe invece alcuna variazione di
temperatura.
Famiglia di elementi chimici tutti gassosi a temperatura ambiente e che comprende, in ordine di peso atomico crescente, l'elio, il neo, l'argo, il cripto, lo xeno e il rado. Il nome di g. rari, usato soprattutto in passato per indicare questi elementi, era in realtà poco giustificato perché essi sono contenuti nell'atmosfera in percentuale assai diversa dall'uno all'altro e che per il più abbondante, l'argo, si avvicina all'1%; più giustificato è invece il nome di g. nobili, che indica la loro grande inerzia chimica. Le quantità di neo contenute nell'atmosfera sono molto minori, mentre minime sono quelle di elio, di cripto e di xeno e infinitesime sono quelle di rado, g. fortemente radioattivo che prende origine dalla disintegrazione spontanea dell'atomo del radio. Di origine radioattiva è, almeno in gran parte, anche l'elio atmosferico, contenuto spesso, in percentuali che possono raggiungere anche il 15%, nei g. naturali. Tutti gli altri g. nobili si rinvengono invece in percentuale apprezzabile solo nell'atmosfera. L'inerzia chimica dei g. nobili è dovuta alla stabilità del sistema di otto elettroni che tutti presentano nello strato più esterno del loro atomo (a eccezione dell'elio che ha solo due elettroni nel primo strato). Di conseguenza, essi non tendono a modificare tale sistema per formare legami chimici neppure con altri atomi dello stesso elemento, ossia a formare, p. es., molecole biatomiche come quella O dell'ossigeno, e risultano invece costituiti da atomi liberi. Fino al 1962 non era noto alcun composto chimico dei g. nobili, dopo di allora si sono preparati vari composti del cripto e dello xeno con il fluoro, il cloro e l'ossigeno, peraltro tutti difficili da ottenere e più o meno instabili. A formare i legami chimici di tali composti intervengono da parte del g. gli elettroni dello strato immediatamente sottostante a quello più esterno; dei g. nobili più leggeri non è invece noto alcun composto. Anche le forze attrattive di tipo fisico che si manifestano tra gli atomi di un g. nobile allo stato liquido e allo stato solido sono molto deboli e ciò ne spiega il basso punto di ebollizione. Caratteristica dei g. nobili è inoltre la vicinanza del punto di ebollizione e del punto di fusione, per cui tutti possono sussistere allo stato li quido solo in un intervallo di temperatura assai ristretto. I g. nobili si ottengono in genere come sottoprodotto della preparazione dell'azoto e dell'ossigeno puri dall'aria liquida. Fa eccezione l'elio che si ricava invece dal frazionamento dei g. naturali.
L'analisi dei g. può essere effettuata attraverso i tradizionali metodi di assorbimento o, più modernamente, mediante metodi strumentali più sofisticati. I primi sono usati quasi esclusivamente per ricerche di laboratorio, mentre i secondi, oltre che per la ricerca pura, vengono vantaggiosamente usati su scala industriale per seguire le reazioni o per rivelare eventuali perdite nelle apparecchiature. Nell'analisi per assorbimento, il campione di g., il cui volume deve essere preventivamente misurato a una determinata temperatura e pressione, viene portato a contatto con una sostanza, solida o liquida, capace di assorbire o di reagire con uno o più componenti del g.; successivamente si misura la diminuzione di volume del campione. La sostanza usata per l'assorbimento deve essere specifica per ogni costituente della miscela gassosa. In pratica non è possibile trovare per ogni g. un assorbitore specifico, così, p. es., l'idrossido di potassio, comunemente usato per assorbire l'anidride carbonica, assorbe anche altre sostanze gassose acide. In questi casi i componenti della miscela vengono prima ossidati e il loro volume viene determinato dopo l'assorbimento dei prodotti di ossidazione. L'analisi con metodi strumentali si compie mediante analizzatori automatici che, sfruttando le proprietà fisiche dei g., permettono di separare con elevata selettività e di identificare i diversi costituenti del campione. Le proprietà fisiche utilizzate sono la densità, la temperatura, l'indice di rifrazione, la suscettività magnetica, il calore di combustione e di reazione, ecc., mentre gli strumenti usati vanno dai termometri di precisione agli spettrometri di massa, ai gascromatografi, agli spettrofotometri.
S'intendono per tali sia le miscele di g. utilizzate per la produzione di energia termica, che possono essere di origine naturale o essere ottenuti da altre sostanze con processi vari, come i g. di distillazione (g. di città; g. di cokeria, ottenuto come sottoprodotto della cokizzazione), i g. di gassogeno (g. d'acqua; g. d'aria; g. misto), g. liquidi, sia altre miscele gassose com bustibili rappresentate dai sottoprodotti di processi metallurgici (g. d'altoforno, g. d'arrostimento, ecc.) oppure derivati dalla lavorazione termica di prodotti petroliferi (g. d'olio; g. di reforming, ecc.).
|
Privacy |
Articolo informazione
Commentare questo articolo:Non sei registratoDevi essere registrato per commentare ISCRIVITI |
Copiare il codice nella pagina web del tuo sito. |
Copyright InfTub.com 2025