|
|
| |
a1-LITICI
Appartengono a questo gruppo principalmente la fenossibenzamina, la fentolamina, i derivati dell' ergot, prazosina, terazosina, doxazosina.
Sono tutti antagonisti competitivi tranne la fenossibenzamina.
Fentolamina e derivati dell' ergot sono non selettivi; prazosina, terazosina e doxazosina sono invece selettivi.
prazosina agisce come a1-antagonista, in modo diretto sulla parete vasale, competendo col Ca ed inibendo le PDE. Il primo resta comunque l' effetto fondamentale. Non modifica il FPR nè la filtrazione. Interessante l' effetto metabolico di aumento delle HDL e diminuzione significativa delle LDL e dei trigliceridi. Non modifica la liberazione di insulina, nè la glicemia, nè la concentrazione di acido urico (per questi motivi risulta il farmaco di scelta per i pazienti ipertesi iperlipidemici, diabetici o gottosi).
Farmacocinetica: biodisponibilità 60%, soggetta a metabolismo di primo passaggio. 97% legato alle proteine plasmatiche. Eliminazione renale.
Indicazioni terapeutiche:
Ipertensione lieve/moderata/grave: da sola o in associazione a altri antipertensivi (diuretici tiazidici o b-bloccanti). Poichè la prazosina non compromette la funzionalità renale se ne preferisce l' uso in pazienti con IR.
Effetti collaterali e interazioni: tipica l' ipotensione ortostatica dopo la prima dose. Per evitarlo si possono assumere inizialmente dosi basse la sera prima di andare a dormire. Altri EC sono tachicardia, insonnia, nausea, cefalea.
altri farmaci
Terazosina: analoga alla prazosina per meccanismi d' azione e per effetti (compresi gli effetti sui lipidi plasmatici) ma la riduzione pressoria è più graduale e non determina tachicardia riflessa. Il cibo non ne influenza l' assorbimento. E' indicata nell' ipertensione lieve/moderata. Può determinar ipotensione da prima dose, sonnolenza, cefalea.
Fentolamina: vasodilatatore diretto raramente usato nella terapia del feocromocitoma. EC: liberazione di H, blocco canali del K, blocco recettori 5HT ed effetti simili alla fenossibenzamina. T ½ 2 h. Via ev.
Fenossibenzamina: inibitore irreversibile del reuptake di tipo 1 (NA, H, 5HT, Ach) nella terapia del feocromocitoma. EC: ipotensione, arrossamenti, tachicardia, congestione nasale, impotenza. Buona biodisponibilità, t ½ 12 h.
a2 AGONISTI
Agiscono stimolando:
I recettori a2 post-sinaptici di neuroni inibitori a livello centrale (ae nucleo del tratto solitario, centro vasomotore, nucleo del vago) con conseguente ipotensione e bradicardia (quest' ultima soprattutto per aumento del tono vagale)
I recettori a2 presinaptici a livello centrale.
I farmaci più comunemente utilizzati appartenenti a questo gruppo sono la clonidina, il guanabenz, la guanfacina e la metil-DOPA.
Clonidina: Attività ipotensivante associata a bradicardia sinusale e sedazione. Agisce preferenzialmente con la stimolazione di recettori a2 postsinaptici in strutture specifiche con conseguente ipotensione ed aumento dell' attività vagale. Probabile anche l' azione su recettori presinaptici. La clonidina influenza anche la liberazione di altri neurotrasmettitori ed in particolare aumenta il rilascio di oppioidi e di GABA, cosa questa che potrebbe spiegare l' attività sedativa ed anticonvulsivante. Diminuisce i livelli di vasopressina. A livello periferico sembra sensibilizzare i barocettori, contribuendo così all' ulteriore inibizione del simpatico.
Sul cuore la clonidina ha diversi effetti:
inotropo negativo (soprattutto con alti dosaggi)
ridotto output cardiaco (ipotonia simpatica+ipertonia vagale+ridotto ritorno venoso)
A livello circolatorio riduce le resistenze periferiche, preserva il FPR e la filtrazione, migliora il 242i81c circolo cerebrale e ha effetto antianginoso.
Per quel che riguarda la funzione renale si assiste ad un aumento della diuresi per diminuito rilascio di vasopressina, ma tale aumento della diuresi è transitorio.
Farmacocinetica: Biodisponibilità 70-100%, 30% legata alle proteine plasmatiche. Emivita, dopo somministrazione orale, di 6-15 ore. L' effetto ipotensivante si instaura dopo 2-4 ore dalla somministrazione orale e perdura per 7-10 ore. Si può somministrare anche ev e per via transdermica (induce minori effetti collaterali). Viene escreta per un 50% immodificata, per un 50% metabolizzata a livello epatico. L' escrezione è prevalentemente urinaria.
Indicazioni terapeutiche:
Ipertensione lieve/moderata: da sola o in associazione a altri ipertensivi (in particolare diuretici tiazidici o vasodilatatori). Soprattutto ipertensione sistolica isolata.
Crisi ipertensive non da feocromocitoma: infusione ev lenta (altrimenti peggiora l' ipertensione, avendo l' infusione ev un effetto bifasico prima di ipertensione per stimolo dei recettori periferici postsinaptici e poi di ipotensione).
Emicrania (profilassi)
Sindrome da astinenza da oppioidi
Sedazione preoperatoria
Glaucoma
Vampate di calore in menopausa (transdermica)
(bambini iperattivi)
Effetti collaterali e interazioni: i più importanti effetti collaterali (in ordine di frequenza) sono:
sedazione/sonnolenza
xerostomia
vertigini
impotenza
rushes cutanei
ipotensione ortostatica
iperglicemia/intolleranza al glucosio
L' effetto ipotensivante può scomparire con cosomministrazione di antidepressivi triciclici e a-litici (ae yohimbina).
Molto importante sospendere la terapia a scalare lento, altrimenti si rischia una ipertensione di rimbalzo.
altri a2-agonisti
Guanabenz: l' azione è analoga a quella della clonidina, ma ha meno effetto sul cuore (meno bradicardia, minor riduzione della gittata, minor inotropismo negativo...) e non induce intolleranza al glucosio. Riduce inoltre il colesterolo LDL. Escrezione con le urine.
Guanfacina: l' azione è meno intensa rispetto alla clonidina ma più prolungata. Escrezione con le urine (50% immodificato)
Metil-DOPA: l' a-metil-DOPA viene convertita in a-metilnoradrenalina che si comporta come falso neurotrasmettitore. Spiazza infatti la noradrenalina dalle vescicole interferendo così con il rilascio di catecolamine e livello centrale. Si suppone che il farmaco agisca in modo analogo alla clonidina, con stimolazione dei recettori a2 centrali, diminuendo così la pressione arteriosa ed il tono simpatico. L' effetto ipotensivante è dovuto ad una diminuzione delle resistenze periferiche senza modificazione significativa della FC e della gittata. Escrezione con le urine. EC: sonnolenza/sedazione (precoce, dose-dipendente), depressione, xerostomia (precoce, dose-dipendente).
b-BLOCCANTI
b NON SELETTIVI
ALPRENOLOLO
PROPRANOLOLO: antiaritmico di classe 2. Usato nell' angina, IMA, ipertensione, tachiaritmie. 3-5 h. pot 1
OXPRENOLOLO: v. propranololo,
TIMOLOLO: nel glaucoma, nella tireotossicosi e nei tremori da stati ansiosi. 4-5 h
PINDOLOLO: AP, antipertensivo in pazienti con riserva cardiaca bassa e tendenza alla bradicardia. 3-4 h. pot 5-10.
LABETALOLO: è anche antagonista a1 e AP b2. Viene dato nell' ipertensione gravidica.
CARVEDILOLO: è anche antagonista a1
b1 SELETTIVI
ATENOLOLO: nell' angina stabile, ipertensione, aritmie. Controindicato nella bradicardia e nello scompenso cardiaco. Idrosolubile emivita 6-8 h
METOPROLOLO liposolubile emivita 3-4 h
ACEBUTOLO
BISOPROLOLO
b2 SELETTIVI
BUTOSSAMINA
Per la maggior parte di essi l' attività intrinseca è =0, comportandosi come antagonisti competitivi (spostano, cioè, la curva dose-risposta a destra). Alcuni, invece, sono AP ma tale attività si esplica solo in assenza dell' agonista. Principali effetti dei recettori b
b1: cronotropi positivi, inotropi positivi, aumento della velocità di conduzione NAV, aumento del rilascio di renina, aumento della produzione di umor acqueo.
b2: rilassamento muscolatura liscia (vasi, bronchi, utero), vari effetti sul metabolismo (aumento dei livelli di insulina...), aumento dell' apporto ematico al muscolo scheletrico e del suo trofismo.
Una prima distinzione fra i b-bloccanti può esser fatta sulla base della loro selettività recettoriale e dell' attività simpaticomimetica intrinseca.
selettivita' recettoriale: Propranololo, timololo, pindololo, labetalolo e carvedilolo sono non selettivi. Fra i selettivi si possono menzionare l' atenololo, il metoprololo e il bisoprololo (lunga emivita, è attualmente quello più selettivo ed è per ora usato solo ev, invece il betanexolo è disponibile anche per os). La selettività recettoriale risulta vantaggiosa in caso di rischio legato all' inibizione dell' attività b2 (attività broncodilatante, attività metabolica, attività vasodilatante), ma tale selettività si esplica per dosaggi non troppo elevati.
attivita' simpaticomimetica intrinseca: cioè l' agonismo parziale. Tale AP si esplica, come detto, con tono simpatico basso (ae durante la notte). Per cui in condizioni di esercizio fisico, di stress e di altre situazioni caratterizzate da ipertono simpatico l' effetto predominante è quello simpaticolitico. Il prototipo degli AP è il pindololo, che esercita tale AP prevalentemente sui b2.
Un' altra distinzione può basarsi sulle proprietà chimico-fisiche:
farmaci lipofili: hanno come prototipo il propranololo (seguito, in ordine, da alprenololo, bisoprololo, timololo, metoprololo). Hanno un ottimo assorbimento, un alto metabolismo di primo passaggio ed un alto legame alle proteine plasmatiche. Tendono, in pazienti con ridotta funzionalità epatica, ad accumularsi. L' emivita tende ad essere breve (richiedendo quindi più somministrazioni) ed il passaggio della BEE può causare EC al SNC (incubi, depressione, allucinazioni).
farmaci
idrofili: hanno come prototipo l' atenololo. Hanno
assorbimento incompleto, scarso metabolismo epatico e basso legame alle
proteine plasmatiche. L' emivita è elevata (spesso comportano una sola
somministrazione/die). Lo scarso metabolismo epatico li rende adatti a pazienti
con funzione epatica compromessa, ma l' alta escrezione renale li sconsiglia a
pazienti con funzione renale compromessa. Lo scarso passaggio attraverso
farmaci con caratteristiche intermedie: hanno come prototipo il pindololo. Hanno buona biodisponibilità orale e sono eliminati per un 50% circa a livello epatico e per il restante 50% a livello renale.
Poichè molti b-bloccanti (soprattutto quelli non selettivi, primo fra tutti il propranololo) possono dare aumento delle resistenza vascolari periferiche in acuto, sono stati introdotti b-bloccanti con azione vasodilatante periferica tramite antagonismo a1, agonismo b2 o effetti vasoattivi diretti. Così ad esempio il labetalolo ed il carvedilolo, oltre ad essere b-antagonisti non selettivi, sono anche a1 antagonisti. Hanno anche azione b2 agonistica.
Il sotalolo ha la peculiarità di allungare il potenziale d' azione cardiaco oltre che di agire come b-bloccante. Tali caratteristiche lo rendono un buon antiaritmico.
Effetti farmacologici dei b-bloccanti:
effetti
sul cuore: riducono la velocità del potenziale pace-maker
nel NAS (risultando quindi cronotropi negativi), aumentano i tempi di
conduzione a livello del NAV (agiscono sulla depolarizzazione Ca-dipendente)
senza però influenzare i tempi di conduzione intraventricolari, diminuiscono la
gittata (sia per riduzione della FC che per interferenza sulla funzione
contrattile da parte del cAMP che va a fosforilare tramite
effetti sulle resistenze periferiche: gli effetti emodinamici differiscono da acuto a cronico. Tipicamente in acuto la somministrazione di b-bloccanti in pazienti ipertesi provoca riduzione (del 20%) della gittata cardiaca e aumento delle resistenze periferiche (del 20-30%). Dopo tempi variabili da ore a giorni si assiste ad un ritorno delle resistenze periferiche ai valori iniziali ed una stabilizzazione della gittata su livelli più bassi. I farmaci dotati di AP e quelli con proprietà vasodilatanti riducono le resistenze periferiche e diminuiscono meno la gittata. Da notare comunque che, pur con queste differenze, la diminuzione della pressione è analoga nei vari farmaci. Gli effetti sulle resistenze dipendono da due meccanismi fondamentali:
Blocco della vasodilatazione mediata dai recettori b2.
Aumento del tono simpatico, in risposta ad una riduzione della gittata.
I b-bloccanti non esplicano effetti significativi sulle coronarie. Piuttosto, poichè l' attivazione a-adrenergica può causare vasocostrizione a livello coronarico, il b-bloccante può peggiorare tale effetto (ciò è importante il pazienti coronaropatici o con coronarie iperattive ae angina variante).
effetti sulla tolleranza allo sforzo: diminuiscono la tolleranza allo sforzo in maniera dose dipendente. Ciò dipende dalla riduzione della gittata e dalla diminuzione dell' apporto ematico al muscolo per blocco dei recettori b2.Globalmente, cioè, si ha ridotta perfusione periferica.
effetti
sul metabolismo lipoproteico: classicamente i b-bloccanti (in
particolare quelli non selettivi) danno riduzione delle HDL (
Indicazioni terapeutiche: sono estremamente varie:
ipertensione: insieme ai diuretici ed agli ACEI sono gli unici per i quali è dimostrata efficacia nel ridurre la morbidità e la mortalità. Tutti i b-bloccanti hanno uguale efficacia come antipertensivi. E' probabile che l' effetto antipertesivo sia da imputarsi a diversi effetti:
blocco dei recettori b presinaptici facilitanti la liberazione di CA
inibizione della liberazione di renina
diminuzione della gittata e resetting dei barocettori
angina: accanto alla riduzione della sintomatologia riducono anche le probabilità di reinfarto, la morte improvvisa e la mortalità cardiovascolare globale. La loro efficacia nell' angina cronica stabile risiede nella riduzione, da parte del miocardio, del consumo di ossigeno (principalmente per riduzione del prodotto FCxPA e della contrattilità cardiaca) ed un aumento della perfusione per aumento tempo di diastole. Tutti i b-bloccanti hanno uguale efficacia nella terapia della malattia ischemica del miocardio. Sono associabili a nitroderivati e Ca-antagonisti diidropiridinici (tipo nifedipina e amlodipina), essendo anche in grado di ridurre la tachicardia riflessa indotta da queste due classi di farmaci. L' associazione con Ca-antagonisti non diidropiridinici può invece far precipitare la funzionalità miocardica. Alcuni studi mostrano che i b-bloccanti sono efficaci nella riduzione dell' angina instabile in termini di sintomi e di episodi acuti. Inefficace, o addirittura dannoso, risulta essere il trattamento dell' angina variante (effetto "smascherante" sull' attività vasocostrittrice a-adrenergica).
prevenzione dell' ima: in termini di prevenzione primaria non sembrano esserci risultati rilevanti. Il trattamento precoce dell' IMA (entro le prime 72 h) iniziato per ev e proseguito per os ha dimostrato una riduzione significativa della mortalità, di arresto cardiaco e di reinfarto. Anche il trattamento postinfartuale (dopo 72 h) ha dimostrato benefici significativi. Tuttavia, poichè queste terapie vanno continuate per 2 anni, occorre stabilirne l' effettiva validità soggettiva. Fattori positivi sono ae ipertensione ed angina, fattori negativi invece sono broncospasmo, bradicardia e BAV. Si somministrano di solito farmaci selettivi.
aritmie: a parte il sotalolo, tutte le proprietà antiaritmiche dei b-bloccanti sono da attribuirsi al blocco degli effetti proaritmogenici del simpatico. Pertanto hanno particolare efficacia nel trattamento delle aritmie sopraventricolari ipercinetiche provocate da alti livelli di CA (feocromocitoma, ansia...) o da un' aumentata sensibilità miocardica alle CA (tireotossicosi...). Uno degli aspetti principali dell' azione antiaritmica consiste nell' aumento della conduzione a livello del NAV e nel prolungamento della refrattarietà. L' utilità come antiaritmici risiede quindi soprattutto nel trattamento delle aritmie da rientro, nel controllo della frequenza ventricolare in corso di flutter e di fibrillazione atriale e nella sindrome del QT lungo.
cardiomiopatia ipertrofica: è una malattia AD con un' alta incidenza di morte cardiaca improvvisa, caratterizzata da una marcata funzione diastolica e talvolta da un marcato impedimento all' efflusso. Questi farmaci servono a diminuire il gradiente pressorio intraventricolare tramite una diminuzione della scarica simpatica.
insufficienza cardiaca: i b-bloccanti sono di per sè controindicati in questo tipo di patologia. L' utilità del b-blocco deriva dall' evidenza che l' aumento delle CA gioca un ruolo importante nella patogenesi della malattia poichè tale aumento determina una down regulation recettoriale, in particolare a carico dei recettori b1, ed un disaccoppiamento dalle proteine G dei recettori rimanenti (e quindi ipofunzionalità). Si è visto che i b-bloccanti inducono una up regulation dei recettori. Inoltre, come già detto, riducono la richiesta di ossigeno, evitano il sovraccarico di CA e prevengono il sovraccarico cellulare di Ca. Globalmente si è visto quindi che i b-bloccanti, somministrati con cautela, migliorano l' IC. Ancora migliori i risultati con agenti ad azione vasodilatante, come il carvedilolo (farmaco tuttora codificato nella terapia dello scompenso), con analoghi risultati del bisoprololo e del metoprololo. Globalmente quindi si tende, nell' IC, a somministrare a dosi inizialmente molto basse un b-bloccante accanto alla terapia tradizionale dello scompenso (ACEI, diuretici ed eventualmente digitalici) soprattutto se di grado lieve/moderato (NYHA 2 e 3). Al di fuori di questi pazienti l' uso dei b-bloccanti è controindicato.
malattie extracardiache: ipertiroidismo (oltre ai sintomi diminuisce anche la vascolarizzazione della tiroide), ansia, glaucoma, emicrania (b-bloccanti non AP), emorragie da varici esofagee (propranololo).
Effetti collaterali ed interazioni:
Disturbi cardiaci: in particolare bradicardia (in pazienti con disfunzione del NAS) , BAV (specie se associati a altri farmaci antiaritmici come digossina o verapamile) ed IC.
Asma: anche i b1 selettivi
Aumento delle resistenze vascolari periferiche: aggravamento dei sintomi in pazienti con malattie vascolari periferiche (per diminuito flusso ematico al muscolo scheletrico).
Ipoglicemia: prolungano la risposta ipoglicemica all' insulina (per blocco della stimolazione secretiva indotta dai b2) e mascherano la sintomatologia adrenergica da crisi ipoglicemica.
Diabete: se associati a ACEI
Intolleranza allo sforzo
Effetti sul SNC
Impotenza: si pensa ad un aumento della vasocostrizione mediata dagli a1.
Sindrome da sottrazione: iperattivazione del simpatico circa una settimana dopo l' interruzione del trattamento. Ciò è dovuto probabilmente alla up regulation recettoriale, motivo per cui si raccomanda di non interrompere il trattamento in modo brusco.
CLASSIFICAZIONE DI LEZIONE:
NON SELETTIVI
Propranololo
Timololo
Nandololo
B1 SELETTIVI
Metoprololo
Atenololo
Acebutolo
Bisoprololo
Betaxololo
DOTATI DI ISA
Pindololo
DOTATI DI EFFETTI VASODILATANTI
Carvedilolo
Labetalolo
ANTIARITMICI
I farmaci possono essere suddivisi in categorie in base a diversi parametri. La distinzione classica (di Vaughan Williams) vede farmaci di classe:
1: deprimono la fase 0 bloccando i canali del Na voltaggio-sensibili. Il blocco dell' eccitazione è
proporzionale alla FC.
1a: proprietà intermedie fra 1b e 1c. Chinidina, procainamide, disopiramide.
1b: si associano rapidamente dai canali, nel tempo di un battito. Se prima del battito successivo ci
sarà un battito prematuro, esso verrà bloccato. Altrimenti il farmaco si dissocerà. I farmaci di
questo gruppo si legano preferenzialmente ai canali in stato di depolarizzazione. Poichè questo
stato cellulare è tipico delle cellule dopo un infarto questa categoria di farmaci è importante
nelle aritmie post-infartuali. Lidocaina, mexiletina.
1c: l' associazione e la dissociazione ai canali è molto più lenta. Poichè non mostrano preferenza
per uno stato particolare del canale, non hanno specifica utilità post-infartuale. Piuttosto
determinano una riduzione generalizzata dell' eccitabilità (e della conduzione attraverso le
fibre di Purkinje), essendo quindi utili non tanto per i battiti prematuri quanto per le aritmie da
rientro. Flecainide. >QRS (>QT)
2: esercitano un effetto simpaticolitico. Il simpatico può causare extrasistoli e fibrillazioni per via dei suoi effetti sul pace maker e sulle correnti del Ca nei miocardiociti e velocizza la conduzione a livello del NAV. Per cui questi farmaci sono utili da una parte nelle aritmie post-infartuali (che si credono causate da un aumento dell' attività simpatica), dall' altra nelle tachicardie sopraventricolari. Propranololo. >PR
3: rallentano la ripolarizzazione e prolungano la durata del potenziale d' azione. Probabilmente l'
effetto si esplica su canali del K. Sopprimono le aritmie da rientro ed i foci ectopici. Il
prolungamento del potenziale d' azione può avere il paradossale rischio proaritmogenico, e ciò
vale sia per questa categoria di farmaci sia per gli antiaritmici 1a. Amiodarone, sotalolo. >QT
4: bloccano i canali del Ca. Rallentano la conduzione a livello di NAS e NAV. Utili soprattutto
nelle tachicardie sopraventricolari. Verapamil. >PR
Altrimenti possiamo distinguere gli antiaritmici in base ai meccanismi aritmogenici (Sicilian Gambit). I meccanismi aritmogenici sono essenzialmente tre:
Aritmie da alterato automatismo.
Quanto maggiore è il processo di automatismo, tanto maggiore sarà
Aritmie da triggered activity. Sono dovute alla presenza di potenziali postumi precoci e tardivi nella fase 3 del potenziale. Quando tali potenziali sono sufficienti a depolarizzare le cellule adiacenti si hanno extrasistoli o aritmie complesse come la torsade de pointes.
Aritmie da rientro. Il rientro è dovuto alla presenza di circuiti elettrici isolati (generalmente a due vie di conduzione) con una porta di ingresso ed una di uscita. Affinchè il rientro si stabilisca è necessario il blocco unidirezionale ed il rallentamento in una zona del circuito.
In una percentuale elevata di pazienti (5-10%, ancora più elevata se c'è scompenso) i farmaci antiaritmici hanno effetti proaritmogenici. Tali effetti derivano dal prolungamento della fase di ripolarizzazione (Ia e III), dallo sviluppo di early after depolarizations (digossina) e dalla modificazione, in senso aritmogenico, di un circuito di rientro (un farmaco aumenta il tempo di ripolarizzazione facilitando il circuito; tutti i farmaci). Più spesso gli eventi indotti dai farmaci sono la tachicardia ventricolare, la sindrome del QT lungo e le torsioni di punta e si manifestano a qualche giorno dall' inizio della terapia o dell' aumento del dosaggio.
CHINIDINA: viene utilizzata
per il mantenimento del ritmo sinusale in pazienti con flutter o fibrillazione
atriale e per la prevenzione delle recidive di tachicardia o fibrillazione
ventricolare. La chinidina si lega ai canali del Na allo stato aperto. Col
crescere della dose si avrà blocco prima dei canali del K e poi del Ca. Ha
buona biodisponibilità ed alto legame alle proteine plasmatiche. Metabolismo
epatico. Causa anche blocco a-adrenergico ed ha effetto vagolitico.
PROCAINAMIDE: è un derivato
della procaina. L' attività è simile alla chinidina ma non ha effetto
antiadrenergico e vagolitico. Il suo principale metabolita,
DISOPIRAMIDE: anche questo farmaco ha attività simile alla chinidina ma non ha effetto antiadrenergico. L' effetto antimuscarinico è però presente e responsabile degli effetti collaterali (glaucoma, stipsi, xerostomia, ritenzione urinaria). Può deprimere la funzione ventricolare e causare torsioni di punta.
LIDOCAINA: ev nel trattamento acuto delle aritmie ventricolari, in particolare dopo IMA (infatti è un 1b). La lidocaina blocca i canali sia in stato attivo che inattivo. Poichè il recupero del blocco è molto rapido si spiega come mai sia più attiva in tessuti depolarizzati (come in corso di ischemia) o che presentano una scarica elevata. Alto metabolismo di primo passaggio (meglio quindi la somministrazione ev) con metaboliti poco attivi. EC: una posologia eccessiva in terapia di attacco può dare convulsioni, in terapia di mantenimento si manifestano nistagmo (precocemente), tremori, disartria, capogiri, alterazioni del livello di coscienza.
MEXILETINA e TOCAINIDE: sono analoghi della lidocaina e sono utilizzabili per os a causa del ridotto metabolismo di primo passaggio. Gli EC, essenzialmente tremori e nausea, possono essere ridotti dalla cosomministrazione di chinidina.
FLECAINIDE: derivata dalla procainamide, utilizzata solamente per il mantenimento del ritmo sinusale in pazienti con aritmie sopraventricolari (compresa la fibrillazione atriale). Provoca un aumento marcato del tempo di recupero dei canali del Na. EC: effetto proaritmico, aggravamento dell' insufficienza cardiaca, blocco della conduzione.
SOTALOLO: è un b-bloccante con la peculiare proprietà di bloccare la corrente ripolarizzante del K, prolungando così il potenziale d' azione. Causando un aumento del QT può dare torsade de pointes specie con ipokaliemia.
AMIODARONE: è un analogo dell' ormone tiroideo. Il meccanismo d' azione non è chiaro, interagendo sia coi canali del Na che con quelli di Ca e K. Ha inoltre effetto antiadrenergico. Il risultato, comunque è il blocco dell' attività automatica anomala ed il prolungamento del potenziale d' azione. E' altamente liposolubile ed ha bassa biodisponibilità. EC: data la sua estrema liposolubilità tende all' accumulo causando così fibrosi polmonare (rara alle dosi della fibrillazione atriale) ed accumuli corneali. Altri effetti: disfunzione epatica, tiroidea, fotosensibilizzazione, debolezza muscolare. Nonostante l' allungamento del QT che dà, non ha effetto proaritmogenico.
DOFETILIDE: usata nel mantenimento del ritmo sinusale in pazienti con fibrillazione atriale, è un potente bloccante dei canali del K cardiaci. EC: torsioni di punta (1-3%). Essendo escreta a livello renale si deve porre attenzione alla sua somministrazione in corso di IR. Si utilizza solo in ospedale o comunque sotto controllo strettissimo.
VERAPAMIL e DILTIAZEM: sono
due Ca-antagonisti utilizzati nel controllo della frequenza ventricolare in
pazienti con fibrillazione o flutter atriale. Riducono
DIGOSSINA: usata nel controllo della frequenza ventricolare in pazienti con fibrillazione atriale. E' un cardiotonico che, per la sua stimolazione vagale, da una parte riduce le correnti del Ca a livello del NAV provocandone così la refrattarietà e dall' altra aumenta le correnti del K Ach-dipendenti a livello del NAS provocando così l' allungamento della fase 4 sia nel NAS che nei miocardiociti atriali.
ADENOSINA: bolo ev per il trattamento delle aritmie sopraventricolari. Apre dei canali del K Ach-sensibili. L' azione antiadrenergica di abbassare il livelli di cAMP ha l' effetto di prevenire eventuali DADs. Ha effetto cronotropo negativo, rallenta la conduzione AV. Si somministra in bolo poichè tende ad essere rapidamente metabolizzata da eritrociti e cellule endoteliali. L' emivita è brevissima, di qualche secondo. EC : asistolia <5 sec, vasodilatazione cutanea, dispnea, dolori al torace.
Bloccanti dei Canali del Calcio (Calcio-Antagonisti)
Obiettivo didattico Partendo dalla conoscenza sulla struttura e funzione dei canali transmembrana del calcio (con particolare riguardo ai canali di tipo L) e alla modulazione dei livelli di ioni calcio nel cardiomiocita e nella cellula muscolare liscia vasale, lo Studente dovrà acquisire la conoscenza delle diverse modalità di interazione con i canali transmembrana del calcio e delle diversità di "tropismo tessutale". Dovrà dimostrare la capacità di razionalizzare, sulla base dei concetti precedenti, gli aspetti differenziali di farmacocinetica, di spettro terapeutico, di associabilità con altri farmaci e di effetti collaterali.
Farmaci prototipo: verapamile, diltiazem, diidropiridine di prima (nifedipina) e seconda generazione (amlodipina, lacidipina)
Il Ca a livello vascolare promuove la contrazione attraverso 3 meccanismi:
Apertura dei canali membranari del Ca voltaggio-dipendenti.
Apertura di canali a livello del reticolo sarcoplasmatico ad opera di agonisti che promuovono, tramite secondi messaggeri, la liberazione di fosfatidilinositolo.
Apertura dei canali membranari del Ca attivati da recettori.
All' interno della cellula il Ca si lega alla calmodulina ed il complesso attiva una chinasi per la catena leggera della miosina che, fosforilata, promuove l' interazione fra actina e miosina.
Tutti i Ca-antagonisti rilassano la muscolatura arteriosa ma non hanno grossi effetti su quella venosa, per cui non influenzano tanto il precarico.
a livello cardiaco le correnti che sostengono l' accoppiamento eccitazione-contrazione non sono solo dovute al Ca (come nelle cellule muscolari lisce vasali) ma anche al Na. Il Na passa attraverso canali veloci, il Ca attraverso canali lenti. A livello miocardiocitario il Ca si lega alla troponina, permettendo così l' interazione del complesso acto-miosinico. Per queste ragioni i Ca-antagonisti a questo livello hanno effetto inotropo negativo (clinicamente contrastato dal tono simpatico attivato dai baroriflessi, che tende alla tachicardia ed all' inotropismo positivo). A livello dei NAS e NAV la depolarizzazione dipende invece in larga misura dalle correnti lente del Ca. Per tale ragione i Ca-antagonisti a questo livello agiscono ritardando il recupero del canale lento. Questo è vero più che altro per Verapamil e Diltiazem. Riducono infatti l' entità di Ca che passa attraverso il canale e ne inibisce il recupero, il tutto in maniera FC-dipendente (cioè a FC maggiori si avrà inibizione maggiore), la frequenza pacemaker a livello del NSA e la conduzione a livello del NAV (quest' ultimo effetto è la base per il loro utilizzo nella terapia delle aritmie sopraventricolari).
Amlodipina: lento assorbimento, effetto prolungato. Emivita di 35-50 h. Occorrono 7-10 gg per ottenere l' effetto farmacologico desiderato. Determina una buona vasodilatazione sia periferica che coronarica.
Verapamil: ha effetti vasodilatatori minori rispetto alle diidropiridine. Per l' effetto cronotropo negativo piuttosto marcato che ha, la tachicardia simpatico-indotta viene nascosta.
Diltiazem: provoca un calo notevole sia della FC che della pressione arteriosa. L' effetto inotropo negativo è meno marcato rispetto al Verapamil.
Nifedipina: di per sè sarebbe leggermente bradicardizzante ma dà tachicardia per riduzione della pressione.
La nifedipina a DE50 risulta più affine a canali del CaL attivabili a -15 mV (cellule muscolari lisce), ed è il motivo per cui è miglior vasodilatatore. Verapamil e Diltiazem hanno invece affinità uguale per canali del CaL miocardiocitici, del tessuto di conduzione e del tessuto muscolare liscio, ed è il mitivo per cui sono attivi sia sul cuore che sui vasi.
I Ca-antagonisti determinano un rilasciamento delle arteriole (tutte) ma non delle vene.
Effetti collaterali e controindicazioni: gli effetti collaterali più frequenti sono da imputare alla vasodilatazione e comprendono vertigini, ipotensione, cefalea, vampate (soprattutto con quelli di prima generazione per blocco rapido dei canali), disestesie alle estremità, nausea. Si possono inoltre presentare stipsi (per Verapamile, diarrea per nifedipina), edema periferico (sono anche detti "antipertensivi invernali"), tosse, dispnea ed edema polmonare. Raramente si hanno sfoghi cutanei, sonnolenza ed alterazione dei test di funzionalità epatica.
Il trattamento con Ca-antagonisti è controindicato in pazienti anginosi, peggiorando la patologia di base con l' ipotensione e la ridotta perfusione coronarica; per questo motivo si consiglia la cosomministrazione di un beta-bloccante. E' sconsigliata però la contemporanea somministrazione di Verapamil e beta-bloccante per aumentato rischio di BAV e/o riduzione della funzionalità ventricolare. Il Verapamil si può somministrare nella miocardiopatia ipertrofica.
NITRODERIVATI
Un loro utilizzo nell' IMA può migliorare la contrattilità del ventricolo sn (a basse dosi riduce il danno ischemico, riduce l' area infartuata e diminuisce le complicanze post infartuali), così come un loro utilizzo post-IMA (previene la dilatazione ed il rimodellamento). Sono inoltre utili in condizioni, come l' IC congestizia e la disfunzione ventricolare sn, in cui un trattamento con b-bloccanti e Ca-antagonisti è controindicato.
Essi esplicano la loro azione attraverso una dilatazione dei distretti vascolari con particolare riferimento ai distretti venoso (riduzione del precarico), arterioso (ma a carico dei vasi arteriosi di capacitanza, non di quelli di resistenza; globalmente quindi a dosi non elevate non si avrà riduzione del postcarico) e coronarico (ridistribuzione dall' epicardio all' endocardio del flusso ematico e dilatazione dei circoli collaterali). Si riducono così sia la richiesta di ossigeno da parte del miocardio, sia il lavoro miocardico e migliora la perfusione senza modificazione dei meccanismi di autoregolazione. Sembra che i nitrati abbiano anche un effetto antiaggregante.
I nitroderivati sono di per sè profarmaci e possono funzionare come donatori (nitroprussiato) e generatori (tutti gli altri) di NO. All' interno delle cellule tutti i nitroderivati (tranne il nitroprussiato) subiscono dei passaggi denitrificanti con conseguente rilascio di NO. Esso stimolando la guanilato ciclasi (ne esiste una solubile attivata da NO ed una accoppiata al recettore per l' ANP) determina rilasciamento della muscolatura vascolare per attivazione della PKG che fosforila ed attiva una Ca-ATPasi (estrude Ca).
Il motivo per cui i farmaci agiscono più a livello venoso che arterioso risiede nel fatto che l' azione dei nitroderivati è modulata dall' endotelio quanto più esso in un dato distretto produce NO. Così la produzione di NO a livello venoso è decisamente minore che a livello arterioso. Per quanto però l' endotelio possa operare una modulazione le azioni dei nitroderivati sono indipendenti da esso. Così si ha:
Inibizione dell' aggregazione piastrinica: in condizioni fisiologiche la piastrina attivata rilascia 5HT e ADP, composti in grado di stimolare la produzione endoteliale di NO e quindi la vasodilatazione.
Vasodilatazione: per innalzamento dei livelli di cGMP.
Questi primi due effetti sono amplificati dal dipiridamolo, un inibitore delle PDE (che infatti mantiene alti i livelli di cGMP).
Vasodilatazione coronarica
Inoltre la somministrazione di NO inibisce la produzione di endotelina (un vasocostrittore) e promuove, con meccanismo diretto, la produzione di PGI2 (un vasodilatatore).
Poichè il metabolismo epatico inattiva fortemente questi farmaci si preferisce la somministrazione sublinguale quando si vogliano ottenere risultati rapidi.
In base alla durata d' azione distinguiamo nitroderivati:
A breve durata: nitroglicerina
A lunga durata: isosorbidi
nitroglicerina o glicerina trinitrato (GTN): somministrazione transdermica, sublinguale, orale e ev. Per os è soggetta a forte metabolismo di primo passaggio (bassa biodisponibilità), la somministrazione sublinguale garantisce l' effetto entro 4 minuti, per ev il metabolismo è rapido (fegato, globuli rossi, endotelio e cellule muscolari lisce). I metaboliti sono 10 volte meno potenti del farmaco attivo. L' emivita è di 7 minuti per la somministrazione sublinguale (i metaboliti hanno emivita di 40 minuti). Posologie troppo elevate inducono ipotensione marcata e riducono la pressione di perfusione a livello coronarico (con annullamento dell' effetto positivo sul danno ischemico ed infartuale).
isosorbide dinitrato: è il nitroderivato più utilizzato per os. L' isosorbide mononitrato ha efficacia analoga. L' emivita è di circa 60 minuti.
isosorbide 5 -mononitrato: ha efficacia simile all' isosorbide dinitrato ed ha emivita di 4-5 ore. La biodisponibilità è decisamente migliore.
nitroprussiato di Na: è un vasodilatatore venoso ed arterioso e diminuisce sia precarico che postcarico. Viene rapidamente metabolizzato a cianuro e NO per cui i suoi effetti sono rapidi (viene infatti usato nelle unità di terapia intensiva per la terapia dello scompenso con perfusione renale, coronarica e cerebrale mantenute).
Indicazioni terapeutiche: la principale indicazione terapeutica è la patologia ischemica miocardica. Il meccanismo principale nell' angina non è la coronarodilatazione ma la diminuzione della pressione sistolica (arteriosa e ventricolare), della pressione polmonare e di quella telediastolica.
Angina stabile: i nitroderivati a
rapida azione sono i migliori per gli attacchi acuti (via sublinguale, spray
nasale o transcutanea preventiva). Possono essere assunti anche qualche minuto
prima di un' attività fisica per prevenire il dolore.
Angina instabile: richiede un protocollo terapeutico complesso. In Europa dei nitroderivati si usa l' isosorbide dinitrato.
Angina variante: non riducono la mortalità e l' incidenza di IMA (come invece fanno i Ca-antagonisti).
Ischemia silente: ne diminuiscono l' incidenza. L' isosorbide 5-mononitrato ha outcome sovrapponibile alla nifedipina.
IMA: in particolare GTN ev precocemente ed in piccole dosi. La cosomministrazione di nitroderivati e ACEI è stata convalidata negli studi GISSI-3 e ISIS-4 ed è stata dimostrata una riduzione del rimodellamento. Inoltre i nitrati limitano l' area ischemica. Nell' IMA, i nitroderivati migliorano la perfusione miocardica diminuendo l' area infartuata e contribuendo al recupero del miocardio sia attraverso un meccanismo diretto, che prevede la dilatazione delle arterie coronarie e delle collaterali, la dilatazione delle zone stenotiche e la risoluzione dello spasmo, che un meccanismo indiretto che si avvale di una dilatazione venosa sistemica cui consegue una diminuzione del precarico.
Effetti collaterali e controidicazioni:
Ipotensione: accompagnata da tachicardia riflessa ed aumento della contrattilità miocardica (fattori che aumentano la richiesta di ossigeno da parte del miocardio e dimunuiscono la perfusione coronarica). In tal caso si può utilizzare un b-bloccante. Raramente è grave, soprattutto in pazienti anziani e, somministrati prima dei pasti, in pazienti con ipotensione ortostatica postprandiale.
Cefalea: frequente.
Vampate: frequente.
Debolezza: frequente.
Alitosi: per somministrazioni sublinguali.
MetHb: rara, per dosi elevate.
Crisi nitritoide: cefalea + ipertensione endocranica + anemia + cianosi + ipotensione fino al collasso.
Tossicità da cianuro: nitroprussiato in pazienti con insufficienza epatica o renale o comunque con compromissione epatica (ae insufficienza cardiaca a bassa gittata con fegato da stasi). Si hanno dolori addominali, alterazioni dello stato di coscienza e convulsioni.
I nitroderivati sono controindicati nell' angina da miocardiopatia ipertrofica (aggravano i sintomi), nel cuore polmonare, nei pazienti ipotesi, nell' ipertensione endocranica e nelle anemie.
Tolleranza ai nitroderivati: facilmente indotta da somministrazioni continue o a lento rilascio (efficace la somministrazione transdermica con intervalli di 12 h). Vari meccanismi proposti:
Deplezione di gruppi SH: necessari alla trasformazione dei nitrati a NO.
Attivazione neuroormonale: aumento delle CA, della renina e del SRAA.
Aumento del volume plasmatico: per diminuita pressione capillare.
Down-regulation dei recettori per i nitrati.
Desensibilizzazione della Guanilato Ciclasi.
Obiettivo didattico Lo Studente, partendo dalle conoscenze acquisite sull'organizzazione morfo-funzionale del nefrone, deve acquisire padronanza dei meccanismi differenziali d'azione delle diverse classi di diuretici con particolare attenzione ai gruppi di farmaci utilizzati nel1'ipertensione, nello scompenso cardiaco e nell'edema polmonare, la capacità di razionalizzare le possibili associazioni dei diuretici tra loro e con altri farmaci. Deva inoltre acquisire la conoscenza dei meccanismi di resistenza all'azione diuretica e degli aspetti di tossicità di organo e/o metabolica.
Farmaci: (sottoclassi e prototipi): Diuretici osmotici (mannitolo, glicerolo, urea, isosorbide); Inibitori dell'anidrasi carbonica (acetazolamide) Tiazidici (idroclorotiazide, clortalidone) Diuretici dell'ansa o drastici (furosemide, acido etacrinico) Diuretici antialdosteronici e "risparmiatori di potassio" (spironolattone e canrenone, amiloride, triamterene)
Note di fisiologia: il rene produce 120 ml di ultrafiltrato/min ma il 99% viene riassorbito. Circa il 65% dei filtrati (quindi anche Na, Cl e K) sono riassorbiti a livello del tubulo prossimale. Tale riassorbimento è isotonico, poichè è alta la permeabilità all' acqua. Il segmento discendente dell' ansa è permeabile all' acqua ma poco permeabile a NaCl e urea. Il segmento ascendente invece è impermeabile all' acqua e permeabile a NaCl ed urea, ed è a questo livello che viene riassorbito il 25% del sodio filtrato. Il tubulo distale può riassorbire Na. I dotti collettori sono deputati alla regolazione fine del filtrato, sensibili come sono all' aldosterone ed all' ADH.
I vari soluti possono venire riassorbiti attraverso le cellule (via transcellulare) o attraverso le zonulae occludentes che separano le cellule fra di loro (via paracellulare). Le zonulae occludentes hanno permeabilità estremamente diversa fra zona e zona dei tubuli. Tubulo prossimale:
![]()
![]()
LUME SANGUE
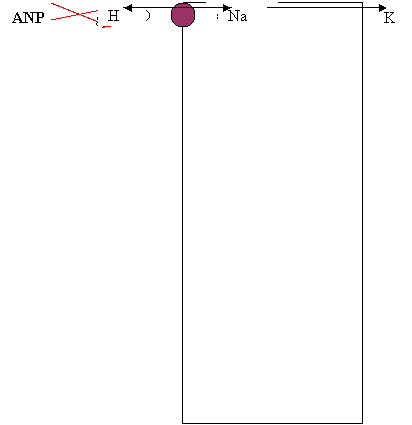

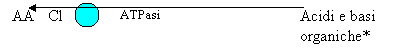
* Acidi: acetazolamide, salicilati, acido urico, diuretici tiazidici, furosemide, indometacina, metotrexato, mdc renali, penicillina, probenecid, gran parte delle penicilline e delle cefalosporine. Basi: amiloride, dopamina, istamina, morfina, serotonina, chinina, atropina.
Ansa di Henle e seguenti: vedi pag 404 e seguenti fig 20.10 e seguenti del Rang Dale
Vedi presentazioni.
Obiettivo didattico: Tale argomento richiede che lo Studente abbia consolidate conoscenze sulla fisiopatologia e biochimica del sistema renina-angiotensina-aldosterone, sui recettori dell'angiotensina II e sulle loro influenze sull'omeostasi cardio-circolatoria e sulla funzione glomerulare; sugli effetti mitogeni dell'angiotensina II sulle cellule dell'apparato cardio-vascolare. Lo Studente dovrà acquisire conoscenza dei meccanismi d'azione differenziali delle due classi di farmaci (inibitori dell'enzima di conversione o ACE-inibitori e sartani) e la capacità di razionalizzazione del loro impiego nelle patologie cardiovascolari. Dovrà acquisire padronanza delle informazioni relative agli effetti sulla mortalità e morbilità da scompenso cardiaco desunto dai risultati dei trials clinici pertinenti. Dovrà possedere la capacità di razionalizzazione dell'associabilità con i diuretici, i concetti relativi alla nefroprotezione e degli aspetti, anche differenziali, di tossicità.
Farmaci (classi e prototipi): ACE-inibitori (captopril, enalapril, lisinopril); Sartani (losartan, valsartan, candesartan)
Il SRAA ha un ruolo importante nella regolazione pressoria sia a breve che a lungo termine, e la sua attivazione è operata da tutti quei fattori che determinano una riduzione pressoria (ae vasodilatatori) od una diminuzione effettiva del volume plasmatico (ae diete iposodiche, diuretici, emorragie, scompenso cardiaco, cirrosi epatica, sindrome nefrosica). La renina è prodotta, accumulata e secreta dalle cellule iuxtaglomerulari a livello dell' arteriola afferente al suo ingresso nel glomerulo. Essa è una proteasi che taglia l' angiotensinogeno in AT1. Gli stimoli al taglio sono sostanzialmente 3:
Via della macula densa. La macula densa è composta dalle cellule colonnari specializzate nella parete del segmento ascendente che decorre fra l' arteriola afferente ed efferente. Una diminuzione nel flusso di NaCl comporta lo stimolo, tramite prostaglandine (prodotte probabilmente dalla COX2, unica COX espressa dalle cellule della macula densa), alle cellule iuxtaglomerulari a rilasciare renina. La riduzione del rilasciamento di renina avviene tramite adenosina, che agisce tramite recettori A1.
Via dei barocettori renali. Quando la pressione a livello dei vasi preglomerulari diminuisce si assiste ad un aumento di secrezione di renina. Il primo stimolo è ritenuto essere una distensione dell' arteriola afferente. Si pensa che i mediatori siano le prostaglandine.
Via dei recettori beta adrenergici. Si attivano i recettori beta1 a livello delle cellule iuxtaglomerulari e si ha secrezione di renina.
La secrezione di renina è diminuita da due sistemi a feedback, uno rapido ed uno lento. Il feedback negativo rapido si attua perchè l' AT2 va ad agire sui recettori del sottotipo 1 posti a livello delle cellule iuxtaglomerulari. Il feedback negativo lento si attua invece perchè l' AT2, provocando un aumento della pressione, riduce il tono simpatico renale, aumenta la pressione a livello preglomerulare e riduce il riassorbimento di NaCl a livello del tubulo prossimale (dando quindi ipertono a livello della macula densa).
L' AT1 di per sè ha scarsa attività ed è convertita attraverso l' ACE in AT2. L' ACE è un enzima di membrana posto sulle cellule endoteliali ed è particolarmente abbondante a livello polmonare. L' enzima non converte solo l' AT1 ma inattiva anche la bradichinina ed altri peptidi vasodilatatori.
Per quanto riguarda l' AT2 essa va ad agire su due recettori:
Sottotipo 1: presenta affinità
specifica per il losartan ed è collegato a proteine G. Una via prevede una Gq
che, attivando
Vasocostrizione generalizzata (++ arteriole efferenti del rene).
Aumento del rilascio di NA.
Riassorbimento di Na a livello del tubulo prossimale.
Secrezione di aldosterone.
Crescita cellulare a livello vasale e ventricolare sinistro.
Sottotipo 2: non si conosce esattamente la via di segnalazione ma il recettore di questo tipo è accoppiato ad una proteina Gi.
L' AT2 provoca delle risposte pressorie rapide e delle risposte lente. Le risposte rapide consistono in un innalzamento immediato della pressione per aumento delle resistenze periferiche ma non influenza significativamente la gittata cardiaca, visto che la risposta ipertensiva attiva il riflesso dei barocettori con conseguente aumento del tono vagale e diminuzione del tono simpatico (anche a livello renale, v. sopra). La vasocostrizione è mediata da un effetto diretto (costrizione delle arteriole precapillari tramite recettori di tipo 1, tale costrizione è scarsa a livello muscolo-scheletrico, cerebrale e polmonare) ed uno indiretto (per facilitazione della trasmissione adrenergica). Le risposte lente dipendono invece da una riduzione, nell' arco di giorni, dell' escrezione renale. L' AT2 infatti da una stimola lo scambio Na/H a livello del tubulo prossimale (anche se a alte concentrazioni ha l' effetto paradossale di ridurre il riassorbimento di Na a quel livello), dall' altra promuove la secrezione di aldosterone (che non ha effetti acuti ma coi giorni provoca aumentato riassorbimento di Na e aumentata escrezione di K e H, tutto a livello dei tubuli distali e collettori). per una trattazione sintetica vedi tabella a pag 776 del goodman gilman.
Esistono anche un' AT3, particolarmente attiva nello stimolare la secrezione di aldosterone e la sensazione della sete, ed un' AT4, che provoca il rilascio di PAI1.
Alcuni farmaci modificano la secrezione di renina:
I diuretici d' ansa aumentano il rilascio di renina poichè diminuiscono il riassorbimento di Na.
Gli ACE-I, bloccanti del recettore AT1 e gli antireninici aumentano il rilascio di renina poichè bloccano il feedback.
I FANS diminuiscono il rilascio di renina poichè diminuiscono la produzione di PG.
I simpaticolitici e b-bloccanti diminuiscono il rilascio di renina poichè bloccano la via dei recettori beta adrenergici.
I farmaci attivi sul SRAA sono gli ACEI e gli antagonisti del recettore per l' AT2.
ACEI
L' ipertensione rappresenta uno dei principali fattori di rischio per le malattie cardiovascolari, potendo determinare danni cerebrali, renali, scompenso cardiaco, malattia coronarica, rimodellamento cardiaco (con ipertrofia ventricolare sn). Gli ACEI esplicano i loro effetti antipertensivi attraverso cinque effetti fondamentali:
Inibizione del SRA
Inibizione del SRA tissutale e vascolare (effetto vasodilatante diretto)
Diminuito rilascio di CA dai terminali nervosi
Aumentata formazione di PG e BK
Diminuita ritenzione di Na per diminuita secrezione di aldosterone e per aumentata perfusione renale.
Questi farmaci sono indicati in monoterapia nel trattamento dell' ipertensione di grado lieve e moderato.
La loro efficacia nella riduzione della pressione arteriosa è maggiore se associati a restrizione salina o ad altri farmaci antipertensivi. L' efficacia di questi farmaci è tanto maggiore quanto maggiore è la concentrazione iniziale di renina. Pazienti di razza nera rispondono peggio agli ACEI e pazienti anziani (che in teoria dovrebbero rispondere peggio, dati i bassi livelli di renina) rispondono anche in monoterapia.
Per quanto riguarda i benefici effetti che questi farmaci hanno sul rimodellamento, possiamo dire che sono efficaci sia nello scompenso (in particolare se associati a diuretici) che nell' IMA (dove uno dei maggiori cambiamenti nella funzione ventricolare sn avviene precocemente con progressiva dilatazione ventricolare ed ipertrofia).
|
Privacy |
Articolo informazione
Commentare questo articolo:Non sei registratoDevi essere registrato per commentare ISCRIVITI |
Copiare il codice nella pagina web del tuo sito. |
Copyright InfTub.com 2025