|
|
| |
1. INTRODUZIONE
1.1 Ciclo cellulare
Il ciclo cellulare è una serie ordinata di eventi funzionali per la crescita e proliferazione cellulare.
Il ciclo cellulare è suddiviso in varie fasi (come mostrato in Fig.1):
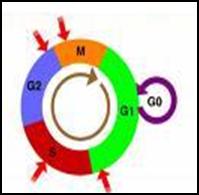
Fig.1 Il ciclo cellulare e i suoi punti di
controllo
Le fasi G1, S e G2 nel loro insieme vengono indicate come interfase, vale a dire il periodo compreso tra una mitosi e quella successiva.
Per passare da una fase all'altra le cellule devono passare dei punti di controllo (indicati in figura con delle frecce rosse), che devono garantire il corretto completamento delle fasi precedenti.
La fase G1 è caratterizzata dalla ripresa di tutte le attività biosintetiche della cellula che si erano arrestate in fase M. Le cellule in questo stato sono caratterizzate da un corredo cromosomico diploide (2n). Le cellule, prima di uscire dalla fase G1, devono superare un punto critico detto punto di restrizione (o START in lievito), nel quale possono arrestarsi (ed entrare in quiescenza in fase G0) se le condizioni ambientali non sono adeguate. In caso di danno al DNA, la proteina p53 è rapidamente indotta e determina l'arresto del ciclo cellulare al punto di controllo in G1, permettendo la riparazione del danno, prima dell'entrata della cellula nella fase S.
Alla fase G1 segue la fase S in cui avviene la sintesi di DNA e l'intero corredo cromosomico viene duplicato e vengono sintetizzati gli istoni.
Una volta terminato il processo di replicazione la cellula ha un corredo cromosomico tetraploide (4n) ed entra in fase G2. A questo punto si innescano meccanismi di controllo dell'avvenuta neo-sintesi, e riparazione del DNA, nel caso di non corretta sintesi, prima che sia permessa la divisione cellulare.
La transizione dalla fase G2 alla profase non è definita chiaramente, comunque una cellula è generalmente considerata in profase quando i singoli cromosomi si sono condensati fino al punto da essere visibili come entità discrete al microscopio a luce trasmessa e ogni cromosoma è formato da due cromatidi fratelli, strettamente legati tra loro in una regione di costrizione, detta centromero (1).
L'inizio della prometafase è segnato dalla disgregazione dell'involucro nucleare, che si riduce a una serie di piccole vescicole membranose, mentre i centrosomi si muovono verso i poli opposti della cellula, iniziando la formazione del fuso mitotico; i microtubuli del fuso prendono contatto con particolari sequenze di DNA, dette CEN (centromeriche) localizzate nei centromeri di tutti i cromosomi e alle stesse si legano un gruppo di proteine specializzate che formano un complesso DNA-proteine chiamato cinetocore.
Durante la metafase i cromosomi sono nel loro stato di massima condensazione e sono allineati sulla piastra metefasica, sul piano mediano della cellula. Questa fase dura circa 20 minuti dell'ora circa richiesta per l'intera mitosi.
Durante l'anafase i cromatidi fratelli si separano e si spostano verso i poli opposti dell'apparato mitotico, o fuso, ed ognuno segrega in una delle due cellule figlie. Questa fase dura in genere pochi minuti ed è la più corta dell'intera mitosi.
Nella telofase si ha la formazione del nuovo involucro nucleare nelle cellule figlie, la decondensazione dei cromosomi e la citochinesi, che dà luogo a due cellule figlie in interfase.
Anche l'apparato del Golgi e il reticolo endoplasmatico si frammentano in vescicole, durante la mitosi, e si riformano nelle due cellule figlie, dopo la divisione cellulare.
1.2 Il controllo del ciclo cellulare.
Le molecole che guidano la progressione del ciclo cellulare, attraverso i vari stadi, sono una serie di complessi proteici costituiti da due subunità: una catalitica, cinasi-ciclino-dipendente (Cdk) e una regolatrice, la ciclina. Il termine ciclina è dovuto al fatto che queste proteine vanno incontro, nel corso del ciclo cellulare a rapidi cicli di sintesi e degradazione.
Le cicline contengono una sequenza estremamente conservata all'estremità N-terminale, chiamata cyclin-box, con la quale esse sono in grado di legare le Cdk, ed una specifica sequenza PEST (Prolina, Glutammato o Aspartato, Serina, Treonina), all'estremità C-terminale. Questa sequenza è bersaglio di ubiquitinazione che indirizza al proteasoma, al momento in cui non sono più necessarie.
Le cicline coinvolte nella regolazione del passaggio de G2 a M sono chiamate cicline mitotiche e le rispettive Cdk cui si legano, nella stessa fase, sono dette Cdk mitotiche; in modo analogo abbiamo le cicline G1, coinvolte nel passaggio di fase tra G1 ed M, cui si legano le Cdk G1.
Al contrario delle cicline, la concentrazione di Cdk rimane costante durante tutto il ciclo cellulare e la loro regolazione dipende sia dalla concentrazione di cicline, sia da un meccanismo di fosforilazione.
La progressione attraverso il punto di restrizio 717d37h ne in G1 è controllata dai complessi Cdk4, e Cdk6 con le cicline di tipo D. La transizione dalla fase G1 a S è mediata dalla fosforilazione del principale bersaglio del complesso Cdk4-6/ciclinaD e cioè la proteina Rb, una molecola che controlla l'espressione di geni, come le cicline E ed A la fosfatasi cdc25, e la DNA polimerasi α, necessarie per il superamento del punto di restrizio 717d37h ne e per la sintesi di DNA.
pRb esercita questo controllo legandosi e sequestrando il fattore trascrizionale E2F; una volta fosforilato dai complessi Cdk/ciclina, pRb, perde affinità per E2F che è quindi libero di svolgere il suo ruolo (2).
La ciclina E, una volta complessata con la Cdk2, fosforila ulteriormente pRb, completandone l'inattivazione e permettendo la sintesi di altri fattori necessari alla cellula per entrare nella fase S, tra i quali si ha nuova sintesi di ciclina A che a questo punto raggiunge la sua concentrazione massima (3). Il complesso Cdk2/ciclina A è necessario per l'ingresso e per la progressione attraverso la fase S.
Durante la fase G2 abbiamo la formazione di un complesso Cdk1/ciclina A il cui ruolo non è ancora stato completamente chiarito, ma che sembra essere coinvolto nell'inibizione di un inibitore della mitosi.
I complessi Cdk/ciclina mitotici vengono sintetizzati durante le fasi S e G2, ma la loro attività viene inibita fino a quando non viene completata la sintesi di DNA, tra questi il complesso Cdc2/ciclina B spinge la transizione da G2 ad M.
1.3 Inibitori del ciclo cellulare
Gli inibitori del ciclo cellulare si possono suddividere in due famiglie: la famiglia degli INK4 (inhibitors of kinase 4, inibitori della cinasi 4), di cui fanno parte p15, p16, p18 e p19; e la famiglia delle CIP/KIP (Cdk inhibitory protein, proteina inibitoria delle Cdk), cui appartengono p21, p27 e p57 (1).
Le CIP/KIP si legano a tutti i complessi formati da Cdk1, Cdk2, Cdk4 e Cdk6 + ciclina e ne inibiscono l'attività interagendo con entrambe le subunità, e inserendo un'α-elica nel solco catalitico, adibito al legame con l'ATP.
Gli INK4 inibiscono la formazione di complessi Cdk4/ciclinaD e Cdk6/ciclinaD, legandosi stabilmente alle Cdk4 e 6. Un'iperespressione delle proteine della famiglia INK4 porta all'inibizione della progressione del ciclo cellulare e all'arresto in fase G1. Tutte le proteine INK4 contengono quattro ripetizioni in tandem di una sequenza consenso, di circa 32 residui, chiamata ripetizione anchirina, dal nome della prima proteina in cui è stata identificata questa sequenza ripetuta.
p21 svolge il suo ruolo di inibitore dei complessi Cdk/ciclina in risposta ad un danno al DNA. Topi knockout omozigoti per p27 mostrano una crescita superiore del 30%, rispetto a topi normali con le stesse settimane di vita, a causa di una generalizzata iperproliferazione delle cellule, nella maggiore parte degli organi, ma alla fine la maggior parte di queste cellule si arresta in fase G1. Topi knockout per p57, che non sintetizzano affatto questa proteina, muoiono poco dopo la nascita a causa di numerosi difetti nello sviluppo di tessuti in cui normalmente questa proteina è espressa (4).
1.4 Neoplasia
Quando i normali meccanismi di controllo della crescita, e della regolazione del ciclo cellulare, non funzionano, la proliferazione cellulare incontrollata può produrre una massa di cellule chiamata tumore. In base alla loro capacità di diffondere, i tumori sono classificati come benigni o maligni. I tumori benigni sono masse localizzate che non diffondono, mentre i tumori maligni possono diffondere nei tessuti circostanti e nell'intero organismo, conducendolo. Un tumore maligno viene definito con il termine generale di cancro.
La causa principale della perdita dei normali meccanismi di controllo di una cellula è da ricercarsi in mutazioni del DNA, che possono essere spontanee, dovute ad errori durante la replicazione o introdotte nelle cellule da infezioni virali, prodotte da agenti chimici, ecc.
Tutte queste mutazioni, in grado di indurre il cancro, sono comunque a carico di tre classi fondamentali di geni: gli oncogeni, i geni oncosoppressori e i geni coinvolti nella riparazione dei danni al DNA.
Un oncogene è un gene, la cui mutazione è in grado di indurre lo sviluppo di un tumore in maniera dominante: un solo allele oncogenico mutante può alterare il fenotipo cellulare. La forma non mutata dell'oncogene, presente in cellule sane, è chiamato proto-oncogene.
Vi possono essere varie mutazioni a carico di un proto-oncogene: mutazioni puntiformi, riarrangiamenti del DNA (delezioni o scambi di sequenze), amplificazione genica, traslocazione cromosomica.
La maggioranza degli oncogeni codifica per componenti delle vie del segnale mediato da fattori di crescita, quali: i fattori di crescita stessi, i recettori, le proteine G associate alla membrana plasmatica, le proteine cinasi, i complessi Cdk/ciclina.
Un'alterazione di un recettore di un fattore di crescita, per esempio, può causare la permanente attivazione del dominio tirosina cinasico anche in assenza dello specifico ligando legato al recettore.
Un oncogene particolare è rappresentato da Bcl-2, che codifica per una proteina (Bcl-2) che blocca l'apoptosi. In questo caso l'insorgenza del tumore è causata non da un'eccessiva stimolazione proliferativa, ma da un eccesso di Bcl-2 che blocca l'apoptosi di cellule con danni al DNA (1).
Un tumore può anche essere causato dalla perdita di geni oncosoppressori i quali, in condizioni normali, frenano la proliferazione cellulare. La mutazione di un gene oncosoppressore è recessiva: è cioè necessario che, in una cellula diploide, entrambe le copie del gene siano mutate perché la cellula risulti anormale. I principali geni oncosoppressori sono:
il gene Rb, che codifica per la proteina Rb coinvolta nel check-point cellulare tra la fase G1 e la fase S;
il gene p53, che codifica per una proteina chiamata anche "guardiano del genoma" in quanto il suo ruolo principale è quello di indurre l'arresto del ciclo o la morte cellulare in caso di danni al DNA. Una perdita di funzione di p53 può contribuire allo sviluppo di un tumore, in quanto consente la sopravvivenza e la riproduzione di cellule che hanno danni al DNA.
Anche se nella maggior parte dei tumori umani sono state identificate anomalie del gene oncosoppressore p53, questo non significa che la perdita di funzionalità di p53, da sola, possa essere causa di tumore. I tumori umani più frequenti, tra cui anche quello del colon, sono dovuti a mutazioni multiple che causano, sia l'inattivazione di geni oncosoppressori, che la conversione di proto-oncogeni in oncogeni.
Infine, alterazioni dei meccanismi di riparazione del DNA possono contribuire alla produzione di tumori, consentendo l'insorgenza di mutazioni multiple.
In generale, perché si crei un tumore, è necessario che si generi più di una mutazione, nei discendenti clonali di una singola cellula. Queste mutazioni avvengono soprattutto nelle cellule somatiche e si accumulano nel corso del tempo, alimentando la progressione da cellula normale a cellula tumorale (5).
1.5 I recettori tirosin-cinasi
L'attività cinasica è in genere parte integrante della proteina recettore, in alcuni casi però l'attività di recettore e quella cinasica possono essere svolte da due proteine diverse: in questo caso la tirosina cinasi viene detta tirosina cinasi non associata al recettore; tuttavia questa cinasi può legarsi al recettore ed essere attivata dall'interazione del recettore con il ligando, ottenendo un risultato netto molto simile all'attivazione di un tipico recettore con attività tirosina cinasica intrinseca.
Finora sono state identificate 90 proteine tirosin-cinasi, di cui 59 risultano possedere attività recettoriale (6).
I recettori tirosin-cinasici (RTKs) presentano caratteristiche strutturali comuni che comprendono:
un dominio N-terminale extracellulare, in cui si trova il sito di legame per il ligando;
una regione idrofobica transmembrana, costituita da una singola α-elica di circa 25 amminoacidi;
un dominio C-terminale citoplasmatico responsabile dell'attività cinasica e dell'interazione con i trasduttori del segnale intracellulari.
Gli RTK vengono classificati in recettori tirosin-cinasici e serina/treonina-cinasici in base agli amminoacidi da essi fosforilati.
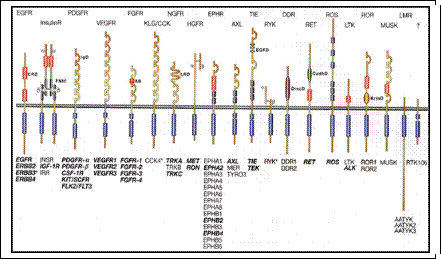
Fig.2: Famiglia dei Recettori Tirosin-Cinasici umani
I recettori tirosin-cinasici vengono suddivisi in sottofamiglie secondo i diversi domini strutturali presenti nella zona extracellulare, che è anche la più variabile, mentre i domini transmembrana e citoplasmatici sono molto più conservati.
La regione C-terminale, una volta attivata dal legame con il ligando, è responsabile dell'interazione con i trasduttori intracellulari del segnale, proteine biologicamente e biochimicamente diverse tra loro, ma con la presenza di domini comuni di legame per le fosfotirosine (PTB) SH2 (Src-homology-2) e domini SH3 che riconoscono sequenze ricche in prolina.
1.6 EGFR
Il recettore per il fattore di crescita epidermale (Epidermal Growth Factor Receptor o EGFR/Erb1/HER1) fa parte della famiglia di recettori con attività tirosina cinasica intrinseca (RTKs) ed è una glicoproteina transmembrana di 170 KDa.
La struttura dell'EGFR comprende un dominio extracellulare glicosilato, con due domini ricchi di residui cisteinici, tra i quali si trova la regione di interazione con il ligando, un dominio transmembrana costituito da una α- elica e un dominio citoplasmatico, sede dell'attività catalitica (7).
La famiglia di EGFR consiste di 4 recettori strettamente correlati: EGFR (Erb1), Erb2 (HER27neu), detto recettore orfano, in quanto non se no conoscono i ligandi, Erb3 (HER3), privo di attività cinasica e attivato dalle erguline, Erb4 (HER4), anch'esso attivato dalle erguline (8).
Le tappe di attivazione di EGFR sono le seguenti:
legame dell'EGF, una piccola proteina monomerica di 53 amminoacidi;
dimerizzazione del recettore, dovuta ad un cambiamento conformazionale, indotto dal legame recettore-ligando, con formazione di complessi omodimerici o eterodimerici con Erb2;
Il dominio intracellulare della proteina cinasi fosforila specifici residui di tirosina presenti sulla coda C-terminale del suo partner di dimerizzazione, in un processo chiamato autofosforilazione o transfosforilazione. Il recettore possiede sei diversi siti di autofosforilazione, tre maggiori (Y1068, Y1148, Y1173) e tre minori (Y 992, Y1045, Y1086) (9).
Reclutamento di proteine adattatrici citosoliche, responsabili della trasduzione del segnale.
A questo punto sono possibili varie vie di trasduzione alternative:
Il recettore in seguito a legame con EGF viene internalizzato per endocitosi, mediante vescicole rivestite di clatrina. E' stato proposto che, all'interno della vescicola il recettore vada incontro a degradazione proteolitica che scinde il dominio citoplasmatico da quello citosolico. Quest'ultimo sarebbe in grado di giungere nel nucleo e di fungere da fattore trascrizione.
In alternativa, l'intero recettore, in seguito all'attivazione da parte di EGF e con essa legata. Può traslocare direttamente nel nucleo. EGFR intero potrebbe essere traslocato al nucleo attraverso un sistema di importo convenzionale, associato con il complesso del poro nucleare (la via Ran/importine), tramite il riconoscimento di una sequenza di localizzazione nucleare (NLS). EGFR si è ritrovato integro nel nucleo ed associato al suo ligando EGF.
Nel nucleo sono stati ritrovati anche i soli frammenti citoplasmatici del recettore, traslocato grazie ad altre molecole.

Fig.3: EGFR nel nucleo
Questi risultati rivestono comunque una notevole importanza in ambito clinico in quanto contribuiscono a spiegare la resistenza dei tumori che ipersprimono EGFR a farmaci che agiscono unicamente bloccando il segnale trasdotto dal recettore (10).
Esaminando il recettore dal punto di vista di un fattore trascrizionale è stata dimostrata: la presenza di un dominio di transattivazione (sequenza C-terminale ricca di prolina) in grado di legare specifiche sequenze promotrici.
In particolare una quota di EGFR nucleare è risultata associata al promotore della ciclina D1, essenziale per la progressione in G1 (11).
L'effetto finale della trasduzione del segnale intracellulare mediato da EGFR, attraverso le varie vie, è la modulazione di segnali di trascrizione a livello del nucleo cellulare che portano a stimolo proliferativo, blocco dell'apoptosi, interferenza con il ciclo cellulare e neoangiogenesi.
L'internalizzazione di EGFR, in seguito al legame con il ligando, è responsabile anche dei processi di riciclaggio e degradazione del recettore stesso. Questi due meccanismi sembrano ligando dipendenti, poiché si è visto che se il ligando è EGF il recettore è per lo più degradato, mentre se il ligando è TGF-α il recettore viene per lo più riciclato. Non si sa se i recettori attivati in membrana, una volta internalizzati, restino attivi fino al momento della degradazione e se i segnali dei recettori internalizzati siano qualitativamente diversi; EGFR rimane comunque tirosin-fosforilato dopo l'internalizzazione e si trova sia legato ad EGF, sia associato a proteine tirosin-fosforilate, negli endosomi precoci (12, 13).
Il percorso è mostrato in figura 4.
Fig.4: Internalizzazione del recettore

1.7 EGFR e le sue mutazioni
L'EGFR è iperespresso e/o costitutivamente attivato in una grande varietà di tumori umani.
Nelle cellule tumorali l'attivazione di EGFR può dipendere:
dall'iperespressione recettoriale che è responsabile della dimerizzazione del recettore ligando indipendente;
dall'emergenza di forme mutate del recettore che ne permettono un'attivazione costitutiva, ligando-indipendente;
dalla presenza di meccanismi ligando-dipendenti eterologhi.
Il meccanismo di iperespressione di EGFR è stato attribuito sia ad attivazione trascrizionale che ad amplificazione genica, anche se, studi più approfonditi, hanno dimostrato che la causa prima sembra essere di tipo epigenetico e cioè un'attivazione trascrizionale.
E' stata riscontrata una iperspressione di ligandi di EGFR in lesioni pre-tumorali, in particolar modo di TGF-α, implicando così questa via di trasduzione autocrina nelle prime fasi della formazione dei tumori. L'iperproduzione di ligandi in concomitanza con l'incremento dell'espressione di EGFR in cellule del cancro facilita lo sviluppo di un pathway di crescita paracrino e/o endocrino.
Il controllo dell'incremento dei livelli di espressione di EGFR nel cancro e l'associazione tra questa iperespressione e il decremento delle possibilità di sopravvivenza nei pazienti hanno portato allo sviluppo di molte strategie terapeutiche che hanno come bersaglio questo recettore, sia in associazione con chiemioterapici, nel caso di tumori allo stadio avanzato, sia singolarmente come terapia preventiva. L'uso singolo di molecole che hanno come bersaglio EGFR, non ha dato però, nei primi test clinici, risultati molto soddisfacenti.
Le mutazioni più comuni di EGFR nei tumori umani risultano essere la delezione di una parte del dominio extracellulare, la quale produce un recettore costitutivamente attivo.
Generalmente la delezione coinvolge le regioni che vanno dall'esone 2 all'esone 7 e più precisamente dall'amminoacido 6 al 273, comprendendo il primo dominio ricco in cisteine, che costituisce il sito di legame per EGF (7). Il ricongiungimento avviene tra l'amminoacido in posizione 5 e quello in posizione 274 e al posto degli amminoacidi deleti si ha la sola inserzione di un residuo di glicina, come mostrato in figura 5 (14).
Questa mutazione è correlata ad un potenziamento della patogenicità e alla resistenza ai chemioterapici. E' conosciuta come EGFRvIII (mutante deleto di 147 kDa) e si ritrova espressa esclusivamente in cellule cancerose dove attiva una classica via oncogenica di trasduzione del segnale.
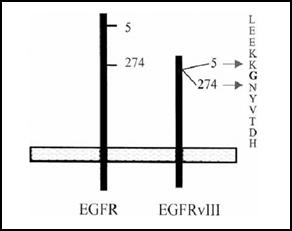
Fig.5: Mutazione di EGFR
L'iperespressione sia di EGFR wild-type che di EGFR mutato è in grado di indurre la trasformazione, anche se è stato visto che appare essere molto più potente quella indotta tramite EGFRvIII rispetto alla forma non mutata (15).
In cellule di tumore del polmone non-small (NSCLC) sono state riscontrate: una delezione dell'esone 19 oppure una sostituzione nell'esone 21 di una leucina con una arginina nella posizione 858 (L858R). Mutazioni in EGFR sono state riscontrate nella maggiore parte dei pazienti con NSCLC responsivi agli inibitori delle tirosin cinasi EGFR (TIKs), quali Gefitinib ed Erlotinib.
I tumori EGFR-dipendenti resistenti ai TIKs presentano generalmente la comune mutazione secondaria in cui la treonina in posizione 790 è sostituita da una metionina (T790M).
La mutazione T790M impedisce l'attivazione di BIM responsabile dell'apoptosi, indotta da Egifitibin. Inoltre, in pazienti che portano la mutazione attivatoria L858R, è stata identificata una seconda mutazione in grado di dare resistenza: la sostituzione della leucina 747 con una serina (L747S), che è in grado di attenuare la up-regolazione di BIM e quindi di ridurre l'apoptosi.
E' stato quindi verificato che BIM (Bcl-2) è l'effettore chiave per l'apoptosi, indotta dai TIKs in tumori causati da alterazioni in EGFR (16).
Attualmente le terapie antitumorali più comuni si basano ancora sull'uso di chemioterapici, la cui azione mira direttamente a danneggiare il DNA o ad inibire la duplicazione cellulare, provocando la morte in maniera aspecifica, con conseguenti danni notevoli per l'organismo.
L'uso di inibitori di TIKs può ovviare a questi inconvenienti ed è in questa direzione che si è orientata una parte consistente dell'attuale ricerca sui tumori.
I due attuali principali TKIs, approvati dalla FDA, sono Gefitibin e Erlotinib.
I TIKs sono derivati sintetici quinazolinici a basso peso molecolare con il seguente meccanismo di azione:
si legano al dominio intracellulare degli RTK;
prevengono l'attivazione della funzione chinasica;
inibiscono il segnale di EGFR (17).
I TIKs bloccano l'attività cinasica di EGFR legandosi al dominio intracellulare in corrispondenza del sito di legame per l'ATP. Attualmente vengono classificati in base al loro grado di specificità recettoriale (azione su uno o più recettori della famiglia ErbB) e in base alla reversibilità o irreversibilità della loro azione. L'irreversibilità è dovuta alla capacità del composto di modificare, attraverso un legame covalente, uno specifico residuo cisteinico a livello del sito di legame dell'ATP. Una loro ridotta efficacia può essere correlata alla presenza, soprattutto in cellule di adenocarcinoma del colon, di mutazioni a livello del dominio cinasico dell'EGFR.
Recenti studi hanno correlato il cambiamento di una singola coppia di basi, nella tasca che lega l'ATP (una metionina al posto di una treonina in posizione 790) con l'acquisizione di resistenza ai TIKs. Il maggior ingombro sterico, conseguente all'introduzione di un residuo di treonina, di maggiore volume, va infatti ad interferire con il legame del ligando (18).
1.8 Naftochinoni e le loro proprietà farmacologiche
I naftochinoni sono composti fenolici diffusi in natura. Sono prodotti del metabolismo secondario di batteri, di funghi così come delle piante superiori. I naftochinoni mostrano delle significative proprietà farmacologiche: sono citotossici e agiscono come antibatterici, antifungini, antivirali, insetticidi, ma possono agire anche come antinfiammatori e antipiretici. Sono conosciuti inoltre effetti farmacologici sul sistema cardiovascolare e riproduttivo. Il loro meccanismo d'azione è complesso: si legano al DNA e inibiscono il processo di replicazione, interagiscono con numerose proteine ad attività enzimatica, infine a livello mitocondriale interferiscono con il trasporto degli elettroni della catena respiratoria. Piante con un contenuto di naftochinoni sono largamente utilizzate in Cina e nei paesi del Sud America dove trovano impiego nel trattamento di neoplasie e di parassitosi (19).
L'effetto antitumorale sembra essere riconducibile alla presenza del nucleo naftochinonico.
L'efficacia delle Shikonine, nel trattamento del cancro, è stata testata anche in vivo utilizzando i topi come modello animale. Gli effetti antineoplastici sono riconducibili alla capacità di inibire la crescita, di indurre apoptosi, e di inibire la DNA topoisomerasi.

Fig 6: Struttura chimica della Shikonina
Numerosi studi hanno dimostrato che un estratto dalla pianta Zicao, il pigmento-L III di naftochinone, è in grado di inibire la proliferazione cellulare di una linea di cellule di cancro dello stomaco e dell'esofago. L'effetto anticanceroso di questo pigmento sembra essere correlato alla sua abilità di influenzare la quantità di RNA cellulare.
Per quanto riguarda l'attività di induzione di morte nelle cellule, β-Hidroxysovaleryshikonin
(β- HIVS), un altro derivato delle Shikonine, è 10 volte più efficace delle Shikonine in varie linee cellulari di tumore. Cellule HL-60 trattate per tre giorni con β-HIVS hanno mostrato in tratti caratteristici delle cellule in apoptosi: la frammentazione del DNA e nucleare, l'attivazione delle caspasi-3, ma non delle caspasi-1, e l'attivazione delle MAP cinasi (quali ERK2 e JNK) e l'attivazione di p38.
Le Dna topoisomerasi sono una classe di enzimi che alterano la conformazione del DNA attraverso una concertata rottura e ricongiunzione delle molecole di DNA, attraverso il controllo dello stato topologico dello stesso (20).
Le topoisomerasi sono coinvolte in molti importanti processi del DNA, quali la replicazione, la trascrizione, la ricombinazione e la segregazione cromosomica. Inoltre le DNA topoisomerasi sono bersaglio di molte molecole antitumorali, che sono già ampiamente utilizzate in terapia.
E' stata riscontrata una forte inibizione della topoisomerasi-1 da parte di derivati dei naftochinoni, sia sintetici che naturali. Dal momento che le shikonine e le alkannine sono naftochinoni naturali e dal momento che l'evidenza ha mostrato che esse sono in grado di inibire la proliferazione di cellule tumorali, questo sono state anche testate per la loro capacità di inibire le topoisomerasi sia in vivo che in vitro. Questi studi hanno dimostrato che i naftochinoni, contenenti almeno un gruppo fenolico idrossile, sono potenti inibitori della topoisomerasi-1. Ciò sembra dovuto all'abilità di queste molecole nel complessare gli ioni di zinco divalenti, correlata con l'attività inibitoria e l'implicita interazione tra le shikonine e il dominio zinc-finger dell'enzima.
In generale comunque questi studi indicano che l'attività delle Shikonine per quanto riguarda l'inibizione dell'attività delle topoisomerasi non è così rilevante come la loro capacità di inibire la produzione di specie reattive dell'ossigeno, questa osservazione suggerisce che la topoisomerasi non è il meccanismo primario dell'effetto anticancerogeno delle Shikonine.
Estratti grezzi dalla pianta di Zicao hanno inoltre proprietà chiemioprotettive contro gli effetti mutagenici indotti dal benzopirene. Queste proprietà delle shikonine e dei suoi derivati sono state testate da numerosi gruppi di ricerca. E' stato dimostrato infatti, che le shikonine, aggiunte alla dieta (0,002 %), inibiscono in maniera significativa l'incidenza dei tumori nei ratti trattati con azoxymetano (un agente mutageno).
Inoltre, in modelli murini, l'angiogenesi indotta da TNF-α ( Tumor Necrosis Factor-α ), viene inibita quando le shikonine sono iniettate assieme a TNT-α (21).
Le Shikonine, isolate da Lithospermum erythrorhizon, sono capaci di indurre apoptosi nella linea cellulare di leucemia premielocitica umana (HL60), inducendo la frammentazione del DNA in multipli di 180 bp e incrementano la percentuale di cellule in apoptosi. L'incremento di cellule apoptotiche è preceduto dall'attivazione della caspasi-3 la quale ha un ruolo centrale nel processo apoptotico.
E' stato dimostrato che le shikonine sono in grado di indurre un decremento dei livelli di fosforilazione di EGFR, ERK 1/2 e di altre proteine tirosin-cinasiche, mentre si nota un incremento nei livelli fosforilazione di JNK1/2 (proteina cinasi stress attivata). Nel complesso, il trattamento con le shikonine è associato ad un incremento dei livelli intracellulari di fosforilazione delle proteine correlate con l'apoptosi e un contemporaneo decremento dei livelli di proteine associate con la proliferazione, questo in particolare nelle cellule di carcinoma dell'epidermide.
Le shikonine inibiscono anche la proliferazione e la migrazione di cellule endoteliali in vitro.
Beta-hydroxyisovalerylshikonin, un derivato delle shikonine è in grado di indurre apoptosi attraverso la modulazione delle MAPKs in una via di trasduzione del segnale Fas-indipendente. Sebbene sia stato dimostrato che derivati delle shikonine inibiscono la fosforilazione delle MAPKs, a tutt'oggi non sono ancora stati identificati i bersagli a monte di questa via. E' stato supposto che le shikonine possano agire da antiproliferativi, attraverso la modulazione del segnale trasdotto da EGFR.
In letteratura e' riportato che un possibile meccanismo d'azione delle shikonine possa riconoscersi nella capacità di inibire la fosforilazione di EGFR. Sia in vivo che in vitro è stato notato che l'attivazione di JNK è necessaria all'induzione di apoptosi; una iperfosforilazione di JNK e' stata infatti ritrovata in cellule trattate. L'apoptosi, prodotta dai chemioterapici, e anche dalle shikonine, in cellule tumorali, sembra essere del tipo Fas-indipendente ed è spesso preceduta da un notevole incremento nei livelli di proteine JNK/SAPK attive (21).
Nel corso della storia estratti provenienti dalle radici della pianta Zicao sono stati anche usati per trattare eruzioni cutanee, mal di gola, bruciature e altro e alle Shikonine sono state attribuite numerose proprietà, oltre a quelle antitumorali: quali proprietà antinfiammatorie, antimicrobiche, antigonadotropiche, anti-HIV-1.
La comparazione tra i vari meccanismi di azione, riportati in letteratura, ha condotto all'ipotesi che il meccanismo di azione delle shikonine possa essere un'inibizione dell'interazione proteina-proteina con numerosi bersagli sia nei comparti intracellulari che extracellulari. Questo generale meccanismo può rendere conto dell'ampio spettro delle attività biologiche e farmacologiche delle Shikonine (22).
1.9 Apoptosi
Il termine "morte cellulare programmata" fu introdotto nel 1964 da Lockshin e Williams (Lockshin e Williams, 1964) basandosi sulla regolarità con la quale specifiche cellule di insetto morivano durante lo sviluppo.
La morte cellulare programmata o apoptosi è una forma di morte cellulare descritta morfologicamente (Kerr et al., 1972) ed è caratterizzata da condensazione della cromatina, frammentazione del DNA e dell'involucro nucleare, formazione di vescicole nella membrana plasmatica, e in ultimo fagocitosi della cellula in apoptosi (Kerr et al., 1991).
L'apoptosi è molto importante nello sviluppo linfocitario, e nell'embriogenesi interviene frequentemente, per esempio, nella rimozione della membrana interdigitale delle mani e dei piedi e durante lo sviluppo di queste; è coinvolta nella morte dei neuroni nella prime fasi embrionali, quando maturano le connessioni nel cervello in via di sviluppo; nell'adulto le cellule del sangue vanno incontro a morte programmata ogni minuto.
La necrosi è invece un processo accidentale ed è il risultato di un danno irreversibile conseguente a:
perdita dell'omeostasi,
danno mitocondriale,
danno esteso della membrana,
fuoriuscita degli enzimi lisosomiali, che causano danno tissutale,
alterazioni nucleari.
L'apoptosi è un processo attivo che richiede consumo di ATP da parte della cellula e che può venire indotto da segnali esogeni (come ad esempio un'infezione virale) o da segnali endogeni, senza conseguenze indesiderate sulle cellule circostanti.
La cellula può entrare in apoptosi in assenza di segnali di sopravvivenza, quali per esempio mediati dall'adesione al substrato o dai fattori di crescita.
L'entrata in apoptosi dipende anche da stimoli di tipo negativo, come l'aumento dei livelli di ossidazione all'interno della cellula, danno al DNA da agenti ossidanti o agenti simili, raggi UV, raggi X, farmaci chemioterapici, molecole che si legano a specifici recettori sulla superficie cellulare, tra cui il Tumor necrosis factor-α (TNF-R1), la Linfotossina (tumor necrosis factor-β), Fas ligand (FasL) una molecola che si lega al recettore di superficie Fas (CD29).
Un ruolo chiave nei processi apoptotici è svolto dalle proteine della famiglia di Bcl-2.
Questa famiglia contiene numerosi membri suddivisi a loro volta tra pro-apoptotici che anti-apoptotici, a seconda del tipo di dominio specifico contenuto da ciascuna: un dominio BH3, appartenente alla categoria delle proteine con funzione pro-apoptotica; o un dominio BH4, caratterizzante le proteine con funzione anti-apoptotica.
I domini BH non hanno attività enzimatica, ma solo funzioni relative alla dimerizzazione.
La formazione di eterodimeri tra anti-apoptotici e pro-apoptotici, porta all'inattivazione di questi ultimi; mentre la formazione di omodimeri di pro-apoptotici, tramite i domini BH3, porta alla formazione di pori sulla membrana mitocondriale esterna.
In assenza di un fattore trofico, la Bad, una proteina solubile pro-apoptotica, si lega alla proteine pro-apoptotiche Bcl-2 e Bcl-xl, che sono inserite nella membrana mitocondriale esterna. Il legame della Bad impedisce l'interazione tra le proteine anti-apoptotiche e la Bax, una proteina pro-apoptotica legata alla membrana.
Conseguentemente si formano canali omo-oligomerici nella membrana mitocondriale esterna, attraverso cui fluiscono ioni con conseguente rilascio del citocromo c, dallo spazio compreso tra la membrana mitocondriale interna e la membrana mitocondriale esterna. Il citocromo c si lega quindi alla proteina di raccordo Apaf-1, che a sua volta promuove la cascata caspasica che porta alla morte cellulare.
In presenza un fattore trofico, come per esempio il fattore di crescita per le cellule nervose (NGF), viene stimolata l'attività della fosfatidil-inositolo 3 cinasi (PI-3 cinasi), la quale a sua volta porta all'attivazione della cinasi Akt che fosforila Bad. Bad fosforilata forma quindi un complesso con la proteina 14-3-3 rimanendo intrappolata nel citosol, così le proteine anti-apoptotiche Bcl-2/Bcl-xl possono inibire l'attività della Bax impedendo il rilascio del citocromo c dal mitocondrio e l'attivazione della cascata caspasica.
L'attivazione delle caspasi è uno degli eventi chiave dell'apoptosi.
Le caspasi sono cisteine proteasi che scindono le proteine in corrispondenza di siti in posizione carbossi-terminale, rispetto a residui di aspartato (Asp-xxx) ad essi adiacenti. Queste proteasi hanno bersagli intracellulari specifici, come proteine della lamina nucleare e del citoscheletro. La scissione di questi substrati porta alla morte della cellula.
La caspasi sono sintetizzate come precursori inattivi, le procaspasi, da cui, attraverso una cascata proteolitica vengono prodotti gli enzimi attivi.
Oltre alla loro già stabilita funzione svolta all'interno delle morte cellulare programmata, ci sono crescenti evidenze che le caspasi contribuiscano anche ad alti processi cellulari. Vi sono un gruppo di recettori chiamati "recettori dipendenti" che regolano la vitalità cellulare in presenza dei rispettivi ligandi; mentre, in assenza dei propri ligandi, questi recettori sono clivati dalle caspasi con conseguente rilascio di frammenti di recettore proapoptotico, o esposizione di death-domain.
Tra questi recettori, bersaglio delle caspasi, è stato identificato anche EGFR.
In termini di trasduzione del segnale il clivaggio dei recettori, mediato dalle caspasi, blocca l'attivazione, dei segnali intracellulari, mediata dai ligandi. E' stato ipotizzato che questo possa essere un altro meccanismo attraverso cui le caspasi innescano la tossicità cellulare e uno shot-down della vitalità cellulare.
EGFR è mediatore di effetti antiapoptotici in varie cellule. Nonostante ciò, diversi studi recenti, hanno dimostrato che, oltre a questa azione antiapoptotica, EGFR, in determinate circostanze, è anche molto importante per l'induzione della morte cellulare. Il complesso che si viene a formare tra EGFR e il recettore CD95 (antigene Fas, APO-1), costituisce il prerequisito per la traslocazione di CD95 sulla membrana plasmatica e l'induzione dell'apoptosi via CD95. L'inibizione dell'attività tirosin-cinasica di EGFR attraverso l'uso di specifici inibitori non ha mostrato effetti sull'eterodimerizzazione del complesso recettoriale, ma è stata in grado di prevenire la fosforilazione di CD95 con conseguente blocco dell'apoptosi. Inoltre, mutanti del recettore CD95 con uno scambio di una fenilalanina al posto della tirosina in posizione 232 e 291, fallivano nella traslocazione alla membrana plasmatica e quindi nella formazione del complesso per la trasduzione del segnale di morte (23).
Il clivaggio di EGFR dipendente dalla caspasi-3 e avviene in corrispondenza di due residui al C-terminale, il principale è un motivo costituito da: aspartato, valina, valina, aspartato (DVVD); il secondario è un motivo: DNPD.
Questo clivaggio, di EGFR, dipendente dalle caspasi, non richiede l'internalizzazione del recettore.
Un clivaggio insufficiente di EGFR mutato risulta in un ritardo, ma non in una prevenzione della morte cellulare apoptotica (23).
1.10 Scopo della tesi
Studi precedenti, hanno evidenziato la capacità della molecola FR18 (2-(8-ammino-actilammino)-[1,4] naftochinone) (B1), di indurre apoptosi in una linea di adenocarcinoma del colon (HT29), interagendo con EGFR, un trasduttore normalmente coinvolto nella traduzione del segnale proliferativo. L'obiettivo di questa tesi è quello di valutare gli effetti biologici di molecole di sintesi come C1 e D1. Le molecole della classe C hanno un core naftochinonico (3 nuclei benzenici condensati), mentre le molecole della classe D sono caratterizzate dal fatto di possedere un core tetracenico (4 nuclei benzenici condensati legate). Le molecole C1e D1 posseggono otto carboni nelle catene laterali.
Il modello cellulare utilizzato e' una linea di adenocarcinoma del colon umano (HT29) e il programma sperimentale e' stato così organizzato:
. valutazione delle dosi citotossiche (IC ) delle molecole, utilizzando il saggio dell'MTT.
valutare degli effetti sulla proliferazione cellulare mediante analisi in citometria a flusso
valutazione delle interazioni di D1 con EGFR mediante analisi in microscopia confocale.
2. MATERIALI E METODI
2.1 Colture cellulari e condizioni di coltura
Le cellule utilizzate per gli esperimenti sono una linea cellulare di adenocarcinoma del colon
HT-29.
Le cellule sono state coltivate in terreno medium RPMI 1640 (Eurobio), arricchito con 10% di FBS (Fetal Bovine Serum, Euroclone) scomplementato a 56°C per 30 minuti, e addizionato con 1% di L-glutammina (Sigma-Aldrich) 2mM.
Le cellule sono state inoculate, alla densità di 1*104 cellule/cm2 in piastre Petri (Orange) o in multiwell da 6 pozzetti, dove crescono in monostrato, e mantenute in incubatore a 37°C con il 5% di CO e a pressione atmosferica. Dopo essere state lasciate aderire per il tempo richiesto, dalla tipologia dell'esperimento svolto, il distacco delle HT-29 avviene mediante tripsinizzazione:
2 lavaggi con tampone fosfato (PBS) costituito da 8 g/L di NaCl (Sigma), 0,2 g/L di KCL (Sigma);
aggiunta di tripsina (Seromed) allo 0,125% in EDTA (Sigma) allo 0,02%, per qualche minuto, in incubatore;
neutralizzazione della tripsina con terreno completo.
Conta delle cellule per mezzo della camera di Burker
C1 e D1 (sintetizzate dal gruppo di ricerca del Professore Melchiorre) vengono disciolte in dimetilsolfossido (DMSO, Sigma-Aldrich) alla concentrazione di 10 mM e mantenute alla temperatura di +4°C.
2.1 Valutazione della dose citotossica (IC ) della molecola, mediante saggio dell'MTT
La vitalità cellulare viene valutata mediante un test che sfrutta una reazione colorimetrica:
l'MTT è un sale di tetrazolio e più precisamente è 3-[4,5-dimetiltiazol-2-il]-2,5-difenil-tetrazolio bromuro. Questo sale viene metabolizzato solo dalle cellule sane attraverso l' azione dell'enzima mitocondriale succinico-deidrogenasi, che lo trasforma nel corrispondente sale di formazano, insolubile in acqua. La quantificazione di questi sali, dopo la solubilizzazione con isopropanolo, fornisce una valutazione indiretta dell'integrità delle cellule in esame. Le cellule vengono inoculate in piastre multiwell alla densità di 1x104 e, dopo il periodo di adesione di 24 ore, trattate in triplo con concentrazioni crescenti delle molecole C1 e D1.
Dopo 24 ore di trattamento, le cellule vengono private del terreno di coltura, addizionate di 200 μL di una soluzione di MTT (0.2 μg/μL in PBS) e incubate per 2 ore a 37°C. La soluzione di MTT viene eliminata e si aggiunge isopropanolo per estrarre i sali di formazano.
Quindi si misura l'assorbanza della soluzione a 590 nm utilizzando lo spettrofotometro (Wallac Victor2 1420 multilabel counter). L'analisi dei dati viene eseguita con il software GraphPad Prism 3.02.
2.3 Analisi del ciclo cellulare al citofluorimetro
Le cellule HT29 vengono inoculate in piastre Petri alla densità di 1*104 cellule/cm2 e, dopo 24 ore di adesione, vengono trattate con terreno con C1 e D1 alle concentrazioni di 1 μM.
Dopo 24 ore di incubazione con il trattamento, si preleva il surnatante, si staccano le cellule adese, tramite tripsinizzazione, e si contano con la camera di Burker. Si effettuano 2 lavaggi in PBS. Le cellule vengono fissate in etanolo freddo al 70% e conservate a -20°C.
La colorazione dei campioni viene effettuata, al momento dell'analisi, con Propidio Ioduro (PI, Fluka) 50 μg/mL e RNasi 10 μg/mL . Si lascia incubare per 30 minuti, a temperatura ambiente e al buio, quindi si esegue la lettura con citofluorimetro Epics Elite (Coulter) dotato di un laser a ioni Argon.
Il ciclo cellulare è analizzato usando i software M Cycle (Verity) e MODFIT 5.0.
2.4 Analisi al microscopio confocale
Le cellule vengono inoculate alla densità di 1×104 cellule/cm² e fatte crescere su vetrini in borosilicato posizionati all'interno di multiwell.
Le cellule sono trattate con D1 alla concentrazione di 1 μM per 1 ora.
Le cellule vengono lavate due volte con PBS e fissate in 500 μL di p-formaldeide al 3% per 15 minuti a temperatura ambiente.
Si effettuano, poi, due lavaggi in PBS-Glicina 0,1M e due lavaggi in PBS/BSA 1% , quindi, si permeabilizzano le cellule con poche gocce di Etanolo freddo al 70% per 2 minuti a -20°C. Le cellule fissate vengono reidratate con due lavaggi in PBS/BSA 1% .
Si utilizza una tecnica di immunofluorescenza indiretta per marcare le cellule.
Le cellule sono marcate con l'anticorpo primario anti-EGFR (0,2 μg/mL) (mAB, Santa Cruz) e incubate per 1 ora in agitazione a temperatura ambiente.
Si effettuano, poi, tre lavaggi in PBS/BSA 1%, quindi, le cellule sono marcate con l'anticorpo secondario coniugato con Alexa 568 per 1 ora in agitazione e a temperatura ambiente.
Le cellule vengono lavate tre volte in PBS/BSA 1% e i vetrini sono prelevati con pinze e montati con Mowiol.
L'analisi è effettuata utilizzando un microscopio confocale MRC1024 BioRad dotato di due laser ed un microscopio Nikon PlanApo.
3. RISULTATI
3.1 Determinazione dell'IC
Per valutare gli effetti citotossici delle molecole D1 e C1 è stato allestito il saggio dell'MTT, come descritto nella sezione "Materiali e Metodi". Sono stati quindi riportati in grafico i valori dell'assorbanza a 590 nm in funzione del logaritmo della concentrazione molare ottenendo una curva sigmoide.
Il valore di IC ottenuto per la molecola C1 è compreso nell'intervallo 10, 7- 19, 8 μM, mentre per la molecola. D1 l'intervallo è tra di 13, 2- 18, 1 μM
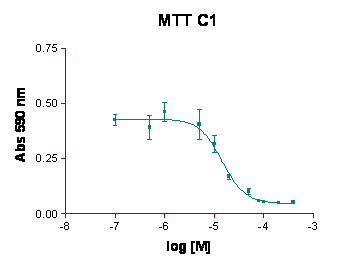
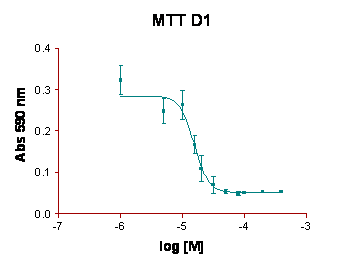
Fig.1: Determinazione dell' IC di C1 e D1 in HT-29 tramite saggio dell'MTT
3.2 Analisi citofluorimetrica del ciclo cellulare
Il profilo di distribuzione, nelle varie fasi, delle cellule trattate con le molecole C1 e D1 è stato analizzato in citofluorimetria a flusso come descritto nella sezione "Materiali e metodi".
I citogrammi riportati in fig.2 rappresentano la distribuzione delle HT-29 nelle varie fasi del ciclo in cellule di controllo, e trattate con una concentrazione di 1 μM delle molecole C1 e D1.
![]()
![]()
![]()
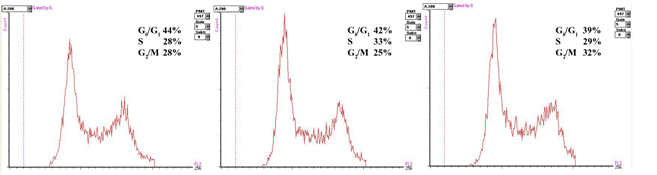
Fig.2: Analisi del ciclo cellulare in cellule HT29: a) popolazione di controllo b) cellule trattate con C1 1 μM; c) cellule trattate con D1 1 μM.
Dai citogrammi possiamo notare che il trattamento con C1 induce una diminuzione delle cellule in fase G0/G1 accompagnata da un aumento di cellule presenti in fase S. Il trattamento con D1 induce una diminuzione ancora più marcata delle cellule in fase G0/G1 che in questo caso si ridistribuiscono in fase G2/M.
3.3 Analisi in microscopia confocale
Le cellule sono state trattate con la molecola D1 ad una concentrazione di 1 μM e marcate con anticorpo anti EGFR come descritto nella sezione "Materiali e metodi". L'immagine ottenuta al microscopio confocale ( Fig.3) mostra come sia presente una colocalizzazione (in giallo) della molecola D1 ( in verde) e dell'EGFR (in rosso).
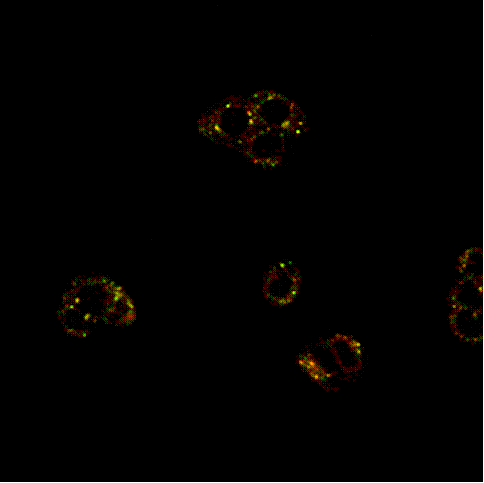
Fig.3 Immagine al microscopio confocale di cellule trattate con D1 ( in verde) e marcate con EGFR (in rosso). Le aree di colocalizzazione sono visualizzate in giallo.
4 . CONCLUSIONI
I risultati ottenuti dal citofluorimetro indicano chiaramente che le sonde C1 e D1, al contrario di FR18, inducono in HT29 proliferazione cellulare.
Qualunque sia il meccanismo coinvolto in questo caso, il risultato da esse prodotto, ci porta ad escluderle totalmente come possibili molecole di interesse terapeutico.
I risultati ottenuti in microscopia confocale, ci hanno invece mostrato una loro interessante caratteristica: colocalizzano con EGFR e questa osservazione le candida come possibili sonde per lo studio di struttura di domini lipidici e del processo di internalizzazione; inoltre, sono sonde che si eccitano ed emettono nel visibile e quindi interessanti per gli studi in cellule vitali.
I microdomini di membrana, conosciuti come caveole, in cui è localizzato EGFR, sono caratterizzati da un'alta concentrazione di gangliosidi, sfingomieline, e colesterolo, oltre alla proteina caveolina. Queste caveole contengono un profilo di proteine altamente specializzate ed escludono la maggior parte delle altre proteine di superficie. Riguardo alla loro composizione lipidica, le caveole sono correlate ai "raft-lipidici" o "cluster -lipidici", altrimenti detti: complessi arricchiti di glicolipidi, insolubili in detergente (DIGS). L'identificazione delle caveole come strutture di membrana, resistenti ai detergenti e contenenti EGFR, è comunque ancora controversa. L'evidenza di recettori caveolari è di tipo biochimico e non morfologico ed è basata su esperimenti di co-sedimentazione delle caveoline e di EGFR su membrane sottoposte a centrifugazione secondo gradiente di densità. La risoluzione di queste frazioni di membrana hanno portato all'identificazione di elevati livelli di EGFR presenti nelle caveole, stimati attorno al 40-60% circa dell'intera popolazione presente sulla membrana plasmatica, considerando una cellula con normali livelli di espressione del recettore.
5. BIBLIOGRAFIA
1. "Il mondo della cellula"
W.M. Becker, L.J. Kleinsmith J. Harding
2. "Retinoblastoma protein recruits histone deacetylase to repress transcription"
A. Brehm, E.A. Miska, D.J. McCance, J.L. Reid, A.J. Bannister, T. Kouzarides; Nature Vol. 391, 5 Feb 1988; 597-601
3. "Functional inactivation of the retinoblastoma protein requires sequential modification by al least two distinct cyclin-cdk complexes"
A.S Lundberg, R.A. Weinberg ; Molecular and Cellular Biology, February 1998, 753-761
4. "Biologia molecolare della cellula "
H. Lodish, A. Berk, S. Lawrence, P. Matsuadaira, D. Baltimore, J.E. Darnell
5. "Genetica. Dall'analisi formale alla genomica".
L.H. Hartwell, L.Hood, M.L. Goldberg, A.E. Reynolds, L.M. Silver, R.C. Veres
6. "The protein Kinase Complement of the Human Genome"
G. Manning et al.
Science 2002; 298: 1-11
7. "EGF receptor".
A. Wells; The International Journal of Biochemistry and Cell Biology 31 (1999); 637-643
"The Epidermal Growth Factor Receptor Family".
L.A. Bazley et al.
Endocrine-Related Cancer 2005
9. "Regulation of Epidermal Growth Factor Receptor signaling by endocytosis and intracellular trafficking"
P. Burke, K. Schooler, H.S. Wiley; Molecular Biology of the Cell, Vol12, June 2001, 1897-1910.
10. "Regulation of Receptor Tyrosine Kinase Signaling by endocytic trafficking"
H.S. Wiley, P.M. Burke; Munksgaard International Publishers, 2001, 2, 12-18.
11. "Inhibition of Autophosphorylation of EGFR by Small Peptides in vitro"
Abeet et al.; British Journal of Pharmacology 2006; 147: 402-411
12. "Quantitative determination of nuclear and cytoplasmatic Epidermal Growth Factor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis"
A. Psyrri et al.; Clinical Cancer Research 2005; 11-16
13. "Nuclear localization of EGFR receptor and its potential new role as a transcription factor"
S.Y. Lin et al.; Nature Cell Biology 2001; 3: 802-808
."Mutant Epidermal Growth Factor Receptors as Targets for Cancer Therapy
Lorimer I.A.J.; Current Cancer Drug Targets, Volume 2, Number 2, June 2002, pp. 91-102 (12), Bentham Science Publishers
15. "Signaling through the epidermal growth factor receptor during the development of the development of malignancy."
J. Rubin Grandis, J.C. Sok; Pharmacology&Therapeutics 102 (2004) 37-46
16. "BIM Mediates EGFR Tyrosine Kinase Inhibitor-Induced Apoptosis in Lung Cancers with Oncogenic EGFR Mutations."
Daniel B Costa, Balázs Halmos, Amit Kumar, Susan T Schumer, Mark S Huberman, Titus J Boggon, Daniel G Tenen, and Susumu Kobayashi; Plos medicine, ottobre 2007
17. "Epidermal Growth Factor Receptor inhibition strategies in oncology"
P.M. Harari et al.; Endocrine-Related Cancer 2004; 11: 689-708
18. "An alternative inhibitor overcomes resistance caused by a mutation of the Epidermal Growth Factor Receptor"
S. Kobayashi et al.; Cancer Research 2005; 65 (16): 7096-7101
20. "Induction of Topoisomerase II-mediated DNA cleavage by the plant of Napthoquinones Plumbagin and Shikonin"
Noboro Fujii, Y. Yamashiata, Y. Arima, M. Nagashima, H. Nakano.
Antimicrobial Agents and Chemotherapy, December 1992, Vol. 36, No.12; 2589-2594
"Shikonin modulates cell proliferation by inhibiting epidermal growth factor receptor signaling in human epidermoid carcinoma cells"
F. Singh, D. Gao, M.G. Lebwohl, H. Wei; Cancer Letters 200 (2003) 115-121
23. "Cellular pharmacology studies of Shikonin derivates"
Xin Chen, Lu Yang, J. Joost Oppenhieim O.M. Zack Howard
Phototerapy Research 16, 199-209 (2002)
24. "Caspase and receptor cleavage"
D. Graf, J.D. Bode, D. Haussinger; Archives of Biochemistry and Biophysics 462 (2007); 162-170
A chi mi è stato vicino
|
Privacy |
Articolo informazione
Commentare questo articolo:Non sei registratoDevi essere registrato per commentare ISCRIVITI |
Copiare il codice nella pagina web del tuo sito. |
Copyright InfTub.com 2025