|
|
| |
Si definisce polimero "una sostanza formata da più molecole dello stesso composto, così che il suo
peso molecolare risulta un multiplo di quello del composto originale".
Catene: lineari, ramificate, reticolate.
Omopolimero, Copolimero (casuale, a blocchi, a innesto).
Polimerizzazione: per addizione (1 reagente, rapida, senza sottoprodotti, poliolefine, vinilici, stirenici), per condensazione (+ reagenti, lenta e incompleta, sottoprodotti (H2O, quindi attento a T alta e troppa H2O se no inverto, riciclaggio oppure effetti indesiderati), poliammidi, poliesteri).
Lunghezza catena: n = grado di polimerizzazione = Mpoli / Monomero.
Massa molecolare media: Mn = ∑nM / ∑n = w tot / N tot
Massa molecolare media ponderale: Mw = ∑wM / ∑w da più importanza alle catene lunghe
Indice di polidispersità: PI = Mw/Mn, se =1 allora distrib stretta, altrimenti varie lunghezze diverse.
Catene non rettilinee a causa rotazione legame sigma, se diluisco singola catena osservo gomitolo statistico il cui diametro è d≈L/50, se considero anche altre catene è la conformazi 959g61j one dello stato liquido: catene raggomitolate e compenetrate senza ordine.
Se raffreddo ho 2 possibilità:
il sistema continua ad essere costituito da gomitoli aggrovigliati, che però perdono in mobilità. Contemporaneamente le proprietà meccaniche diventano simili a quelle di un solido. Il sistema si trova quindi in uno stato di disordine (simile in questo senso ad un liquido), ed in questo senso il polimero si definisce amorfo, e tuttavia le sue proprietà non sono più quelle di un "liquido". Per questo si parla di polimero amorfo vetroso.
le catene possono effettivamente raggiungere delle conformazioni regolari il cui grado di ordine favorisce la minimizzazione dell'energia del sistema: in questo caso le catene danno luogo ad un reticolo cristallino che permette di minimizzare il volume e quindi l'energia del sistema.
Una struttura polimerica completamente ordinata non può tuttavia occupare tutto lo spazio, in quanto il contributo entalpico non prevale sempre su quello entropico legato al disordine e alle irregolarità inevitabilmente presenti nella struttura di una catena polimerica: non esiste quindi la possibilità di avere un materiale polimerico completamente cristallino, ma solamente semicristallino (figura 20), in cui una parte di catene può ordinarsi in un reticolo cristallino, dando luogo ad una fase cristallina che però rimane annegata in un "bagno" di materiale amorfo.
Grado di cristallinità: Xc = Mcristallo/Mtot
Per poter cristallizzare le catene devono accostarsi in modo ordinato (ok iso e sindiotattico, no atattico).
Durante raffreddamento ho bisogno di un certo t per cristallizzare, se Vraff è alta abbasso Xc. Lo stato amorfo non si differenzia da quello liquido se non perché è più viscoso.
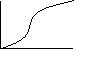
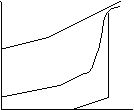
 Comportamento Volumetrico: V V/m
grandezza intrinseca. Per misurare uso bacinella con fluido e immergo il
pezzo (att bagnabilità e porosità), e misuro ΔV al variare di T del fluido
e del fluido col pezzo. Per polimeri amorfi ottengo 2 rette che simbolizzano
comportamento viscoso e liquido. Se osservo il coeff di dilatazione termica a P
cost (α = 1/V [δV/δT]) ho il punto di transizione vetrosa (Tg)
al flesso. Se Vraff è elevata il Volume specifico si discosta dalla linea di
equilibrio ed è maggiore, e non vi ritorna nemmeno se rallento perché non do
tempo di riorganizzarsi a volume min, Inoltre
Comportamento Volumetrico: V V/m
grandezza intrinseca. Per misurare uso bacinella con fluido e immergo il
pezzo (att bagnabilità e porosità), e misuro ΔV al variare di T del fluido
e del fluido col pezzo. Per polimeri amorfi ottengo 2 rette che simbolizzano
comportamento viscoso e liquido. Se osservo il coeff di dilatazione termica a P
cost (α = 1/V [δV/δT]) ho il punto di transizione vetrosa (Tg)
al flesso. Se Vraff è elevata il Volume specifico si discosta dalla linea di
equilibrio ed è maggiore, e non vi ritorna nemmeno se rallento perché non do
tempo di riorganizzarsi a volume min, Inoltre
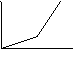
 Volume libero: V = Vocc + Vfree
Volume libero: V = Vocc + Vfree
 Il primo segue andamento lineare e non ha transizione, il secondo dopo
Il primo segue andamento lineare e non ha transizione, il secondo dopo
Pressione: sfavorisce mobilità, Vsi abbassa cosi come
Vfree, che subisce drastica riduzione, Tg aumenta, α diminuisce.
Legame P, Tg: dTg/dP = 0,23 [°C/MPa]
Per abbassare Tg posso inserire nel polimero lubrificanti che aumentano Vfree (CO2, H2O) e mobilità.
Se raffreddo a Vraff cost, dapprima Vfree è suff per riarrangiamenti veloci, fino a che questi non avranno più spazio sufficiente per avvenire. Se Vraff è veloce questa situazione avviene prima: alzo Tg e V specifico è maggiore.
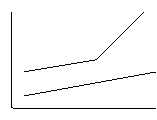
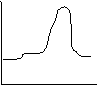 Polimeri semicristallini: parte amorfa si comporta come prima, mentre i
cristalli hanno V pressoché cost fino alla Tm, dove liquefano e V si impenna.
Polimeri semicristallini: parte amorfa si comporta come prima, mentre i
cristalli hanno V pressoché cost fino alla Tm, dove liquefano e V si impenna.
(usa regola della leva per avere % cristallo = Vg-V/Vg-Vc)
Xc diminuisce Vol specifico, mentre Vraff sfavorisce Xc.
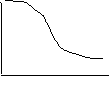
 La cinetica di raff e fus è diversa: la fusione è graduale e avviene in
un campo di T ampio, poiché i cristalli fondono e
La cinetica di raff e fus è diversa: la fusione è graduale e avviene in
un campo di T ampio, poiché i cristalli fondono e
si riformano più perfetti, la mobilità aumenta, la
cristallizzazione avviene a T precise perché perdo mobilità,
cristallizza a T alta e il V si abbassa drasticamente.
Comportamento meccanico: E = F(T,t)/A Lo/ΔL =
σ/ε (grafico E/T)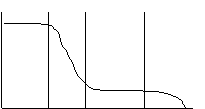 . Noto i tre campi di risposta (Vetro, Gomma, Fluido) e due plateau.
Entrano in campo tre forze: legami covalenti, nodi di reticolazione, van der
waals. Ognuno ha la sua risposta meccanica.
. Noto i tre campi di risposta (Vetro, Gomma, Fluido) e due plateau.
Entrano in campo tre forze: legami covalenti, nodi di reticolazione, van der
waals. Ognuno ha la sua risposta meccanica.
Stretch: risp entalpica (tolgo F torna indietro), modulo E alto per variazione L, basso per rotazione legame.
Sgomitolamento: risp entropica, non è necessaria molta forza perché variano caratteristiche poco energetiche, se sgomitolo troppo arrivo a stretch.
Sgrovigliamento e scorrimento relativo: variazione irreversibile.
Questi tre fenomeni avvengono contemporaneamente, bisogna determinare quale prevale: a T bassa ho poca mobilità, prevale stretch (E alto), dopo Tg gli altri tipi.
Più le molecole sono lunghe più nodi ci sono e scorrimento è difficile, alzo T o aspetto t lunghi, prima della fase fluida. (importante nelle gomme dove è risp prevalente)
Fase vetrosa compresa fra Tmin (infragilimento) e Tmax = Tg - 20°, applicazioni strutturali (per Tg>Tamb). Gomma: solo per applicazioni specifiche, caratteristiche non durano nel tempo.
Polimeri amorfi reticolati: anche a T alte non fluidificano rimangono gomma perché i nodi sono di natura chimica (fino a T in cui si degrada o brucia), ho quindi applicazioni strutturali anche per gomme.
Grado di reticolazione = 1/n° C tra un nodo e l'altro. Se elevato ho E gomma maggiore, inoltre prima di poter sgrovigliare, cosa che avviene per pochi nodi, passo per stretch molecole. Reticolati non riciclabili, macinati e bruciati a T alte per non produrre diossina.
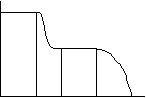 Polimeri semicristallini: cristalli hanno E alto e fragilità, la parte
amorfa a T bassa ha E alto e fragiilità, a T>Tg deformabile, nel primo caso
ho comportamento rigido e fragile, nel secondo rigido e tenace. Dopo Tm no
cristallo fluido E = 0. Se cristalli sono direzionali risp maggiore lungo le fibre. Il
primo plateau è uguale sia per amorfi che per semicristallini, in quanto è la
parte amorfa a rispondere, il secondo è più alto nei semicristallini perché
cristalli conferiscono rigidezza. Xc varia quindi solo il secondo plateau. Se
immetto plastificante anticipo il secondo plateau perché abbasso Tg e posso
effettuare applicazioni a Tamb.
Polimeri semicristallini: cristalli hanno E alto e fragilità, la parte
amorfa a T bassa ha E alto e fragiilità, a T>Tg deformabile, nel primo caso
ho comportamento rigido e fragile, nel secondo rigido e tenace. Dopo Tm no
cristallo fluido E = 0. Se cristalli sono direzionali risp maggiore lungo le fibre. Il
primo plateau è uguale sia per amorfi che per semicristallini, in quanto è la
parte amorfa a rispondere, il secondo è più alto nei semicristallini perché
cristalli conferiscono rigidezza. Xc varia quindi solo il secondo plateau. Se
immetto plastificante anticipo il secondo plateau perché abbasso Tg e posso
effettuare applicazioni a Tamb.
Polimeri: amorfi reticolati (NR, PU), non reticolati (PS ABS, SBS, PMMA, SAN, PC), semicristallini (PP, PE, PA, PETT, PTFE).
Creep e rilassamento: Equazioni costitutive riguardano proprietà intrinseche, che non dipendano da quantità e geometria del manufatto: F σ, ΔL ε; e dalla loro correlazione ricavare C e E. Possono essere semplici o funzione di x, y (accortezza). Comportamento può essere elastico (lineare σ = Eε, non lineare σ = E(ε)ε, ma se tolgo sforzo torno indietro), elastoplastico (tolta F mi rimane deformazione residua), viscoso (τ = η δy/δt). Nei polimeri elastici la deformazione è indipendente dalla storia di sforzo applicata, cosa che non avviene nei plastici. I polimeri sono elastici o viscoelastici.
Prove meccaniche: creep (σ cost), rilassamento (ε cost), sollecitazioni periodiche. Da risp semplici deduci storie complesse.
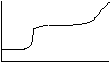
 Creep: ε sale istantaneamente: ho stretch di catena (elastica entalpia,
rigida con legami forti), poi sollecito gomitolo e nodi (risp entropica),
infine i legami tra catene sono meno forti e ho 2 comportamenti: fluido in cui
ε va a ∞, o per i cristalli e i polimeri reticolati rottura o
deformazione lentissima. Cedevolezza: C (t) = ε (t) / σo. Polimeri
viscoelastici lineari: C dipende solo da sforzo; viscoelastici non lineari: C =
C (t,σo). Grafico C/logt: 1° plateau: entalp, 2° entro, poi fluido.
Creep: ε sale istantaneamente: ho stretch di catena (elastica entalpia,
rigida con legami forti), poi sollecito gomitolo e nodi (risp entropica),
infine i legami tra catene sono meno forti e ho 2 comportamenti: fluido in cui
ε va a ∞, o per i cristalli e i polimeri reticolati rottura o
deformazione lentissima. Cedevolezza: C (t) = ε (t) / σo. Polimeri
viscoelastici lineari: C dipende solo da sforzo; viscoelastici non lineari: C =
C (t,σo). Grafico C/logt: 1° plateau: entalp, 2° entro, poi fluido.
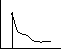 Rilassamento: ho istantaneamente stretch, risp entalpia che accumula stress, che si
rilascia con una risp entropica di sgomitolamento. σ va a 0 nei materiali
amorfi non reticolati (completa riorganizzazione), ≠ 0 nei
semicristallini e reticolati. Modulo di rilassamento:
Rilassamento: ho istantaneamente stretch, risp entalpia che accumula stress, che si
rilascia con una risp entropica di sgomitolamento. σ va a 0 nei materiali
amorfi non reticolati (completa riorganizzazione), ≠ 0 nei
semicristallini e reticolati. Modulo di rilassamento:
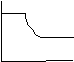 E (t)= σ (t) / εo (Relazione di Hooke) . In scala log vedo 1°
plateau risp entalpica, quindi rilassamento E scende e ho risp entropica, in
seguito scorrimento viscoso. Nache in questo caso ho linearità e non per gli
stessi polimeri di C.
E (t)= σ (t) / εo (Relazione di Hooke) . In scala log vedo 1°
plateau risp entalpica, quindi rilassamento E scende e ho risp entropica, in
seguito scorrimento viscoso. Nache in questo caso ho linearità e non per gli
stessi polimeri di C.
La relazione E = 1/C vale solo all'istante 0, perché il materiale non riconosce istantaneamente la differenza tra un allungamento e uno sforzo impresso in quanto sono vicendevolmente necessari e conseguenti. Dopo ho errore fino al 10%.
Boltzmann: da risp semplici è possibile prevedere sollecitazioni complesse (nell'ipotesi di comportamento lineare). E(t) e C(t) hanno tempi caratteristici diversi.
Postulato di B. : Per un materiale con comportamento lineare la risposta R ad una sollecitazione generica I è pari alla somma di una serie di risposte Ri a sollecitazioni semplici Ii (a gradino).
R(I) = R(I1 + I2) = R1(I1) + R2(I2)
I) pongo prima Δσo a to, poi a t=u pongo Δσ1, ottengo ε(t) = εo(t) + ε1(t) = D(t)Δσo + D(t-u)Δσ2
ε(t) = ∑εi(t) = ∑Δσi D(t-ui)
II) stessa roba per ε.
Variazioni continue: ε = ∫D(t-u) dσ(u) = ∫D(t-u) dσ/du du, analogo per σ.
Prova a velocità di trazione costante: dε/dt = ε° = k ε = kt σ(t) = ∫E(t-u) dε/du du = k ∫E(t-u) du
Vediamo come va E(t) = E∞ + (Eo-E∞)exp(-t/τ) ha due plateau, quindi ottengo
σ(t) = E∞ ε° t - (E∞-Eo) ε° τ [1-exp(-t/τ)],
posto ε = ε° t ho:
σ(ε) = E∞ ε -(E∞-Eo)ε° τ [1-exp(-ε / ε°τ)]
Se aumento velocità k la retta sforzo deformazione è più pendente.
Coefficiente di Poisson: Relazione Cedevolezza a taglio J / Cedevolezza a trazione D:
J = D/2(1+υ)
Sforzi tangenziali: σ = R/s Pa con R raggio e s spessore e Pa la pressione
Flessione estremo libero: f = 1/3 FL3/I C con I per un tubo: π/4 (Re4 - Ri4)
 Struttura e caratterizzazione
dei polimeri
Struttura e caratterizzazione
dei polimeri
Ampie possibilità di conformazioni data libertà di rotazione legame C-C: ad esempio se ho sostituente grosso in posizione isotattica VSEPR crea forma a elica. Per strutture disordinate faccio una descrizione statistica (end to end medium distance, C grado di compattazione).
Polimeri cristallini dispongono catene parallele, quelli amorfi hanno
spazi vuoti, gomitoli e concatenazioni. Il cristallita è piccolo e lamellare, immerso nell'amorfo, con
spessori di 50-
Poi ci sono gli sferuliti (10-100 μm), in cui dal centro partono catene a raggi che si ramificano per mantenere densità di catene costante, e le fibre, fasci di catene parallele, con forti proprietà anisotrope: legami deboli fra catene e forti lungo di esse.
Temperature critiche: Tg, Tm. Prima di Tg ho modulo elevato e fragilità, poi innesco moti di scorrimento viscoso tra catene e il polimero è come un fluido, a modulo molto basso. Tm è propria della parte cristallina, è maggiore di Tg perché i cristalli sono stabili, ma la fusione poi è immediata. Nei polimeri semicristallini tra Tg e Tm ho un comportamento misto, dove i cristallini bloccano gli scorrimenti viscosi della parte amorfa. Tg varia in base a: struttura chimica, configurazione, conformazione, cristallinità, difetti. Tm è influenzata da molti fattori (uno su tutti il PM). Ad esempio se ci sono ponti a H tra catene Tm aumenta, così un nylon 6.6 avrà una Tm maggiore di un PE o di un nylon 10.10, in cui i ponti a H sono più rari. Se ci sono molti legami torsionali C-C favorisco il processo di fusione, e la facilità di rotazione è data dalla presenza o meno di ingombri sterici. Anelli aromatici, ponti ad H e ingombri sulla catena aumentano Tm, legami -O- e -C(=O)- la abbassano. La velocità di raffreddamento massima per ottenere un cristallo è stabilità dall'ingombro sulla catena dei sostituenti, che devono in ogni momento avere il tempo di posizionarsi in modo ordinato per avere la regolarità propria dei cristalli. Se ho doppi legami posso avere conformazione CIS ( che genera forma a elica, molto ingombrante, che formerà cristalli instabili: Tm bassa) o TRANS (struttura regolare, cristalli compatti, Tm elevata). Con operazioni di stiramento il cis può essere allungato (modulo basso) mentre per allungare il trans devo modificare angoli di legame e avrò modulo elevato (se poliisoprene cristallo perfetto E≈88 GPa).
Importanza dei difetti
strutturali: PE: LDPE è amorfo, presenta cristalli
rari e difettosi, mentre HDPE è semicristallino. LDPE ha 60 ramificazioni ogni
Regolarità dei polimeri: chimica (ripetizione di unità, persiste fino alla degradazione del polimero, omoolimero, copolimero), di concatenamento (testa-coda con grossi ingombri cristallizza, testa-testa ovvero irregolarità random con piccoli ingombri, e allora uso ZN), di isomeria geometrica o configurazione (cis, amorfo da elica, trans, cristallino da fibre).
Tassìa: C ibridizzato sp3 è chirale, ma siccome 2 sostituenti su 4 sono i C della catena, non c'è chiralità nel polimero. Piuttosto è utile osservare se tutti i sostituenti sono dallo stesso lato (isotattico), alternati (sindiotattico) o casuali (atattico). In realtà le rappresentazioni grafiche non sono molto veritiere poiché non tengono conto della rotazione di C-C e del fatto che ingombri importanti modificano secondo VSEPR la geometria, ad esempio un polimero isotattico porta tutti i sostituenti all'esterno e da forma a elica.
Polimerizzazione: in base a stechiometria ho poliaddizioni o policondensazioni, in base al meccanismo sequenziale o a stadi. Poliaddizione: (es PE)ho un centro attivo che si propaga al crescere della catena sul nuovo monomero che si unisce (Mn* + M Mn+1*).Policondensazione: (es nylon) casualmente e contemporaneamente reagisce tutto, ho molte catene che crescono insieme ed è una reazione di equilibrio con quindi monomeri non reagiti e prodotti di reazione (in genere H2O). In genere le poliaddizioni sono sequenziali e le policondensazioni a stadi, con delle eccezioni: CH2N2 + CH2CH2 polimero + N2 (policondensazione sequenziale); poliuretani O=C=N-fenolo-N=C=O + HO-CH2CH2-OH -C(=O)-N(-H)-fenolo-N-C(=O)-O-CH2CH2-O- (poliaddizione a stadi, in grassetto il gruppo uretanico).
Polimerizzazioni sequenziali: ho inizialmente l'attivazione della reazione: inserisco un Iniziatore di reazione I che legandosi
a un monomero lo attiva e fa partire la propagazione della catena, che si
arresta quando 2 centri attivi reagiscono (catena morta). Il prodotto finale è
un mix di M singoli, catene morte più o meno lunghe, catene ancora attive
(poche). La lunghezza delle catene non dipende dalla resa della reazione (es PE
ha reazione esotermica ΔH<0 ma con diminuzione di entropia
ΔS<0, quindi per avvenire ΔG = ΔH-TΔS < 0 T< ΔH/ΔS, solo a temperature sufficientemente basse).
Nella polimerizzazione sequenziale ho 3 fasi: l'Iniziazione, con una E di attivazione elevata e una cinetica lenta
(I I* , I*+M M*),
Polimerizzazione radicalica: un radicale è una specie chimica che presenta un elettrone spaiato, non condiviso, ma elettricamente neutra. Esempi: AIBN (azo-bis-isobutilronitrile) CN-C(-CH3)2-N=N-C(-CH3)2-CN rompe legame con i N centrali, che vanno via come N2, e rimangono 2 radicali CN-(CH3)2-C*. Perossido di benzoile: fen-C(=O)-O-O-C(=O)-fen rompe legame perossidico O-O a dare 2 centri attivi -O*. E' caratterizzata da sequenze: Iniziazione, in cui si creano i radicali (I*+M IM*), la propagazione (IM*+M IMM*), e la terminazione per accoppiamento (IM* + *MI IMMI catena morta, nn ho più centri attivi) o per disproporzionamento (R-CH2-C*H2 R-CH=CH2 + H*, H*+H* H2). Ramificazione: Il C* atttivo reagisce con il C lungo la catena, attaccandosi il suo H e lasciandogli il centro attivo, da cui poi parte la ramificazione (LDPE perché c'è minor cristallinità, 60ramif/1000C, HDPE con Ziegler-Natta). La concentrazione di radicali ancora attivi al termine della reazione è dell'ordine di 10^-6 rispetto alle catene di polimeri. Quando ho un monomero reagente con un ingombro sterico (es stireni: CHfen=CH2) il centro attivo si lega al C meno ingombrato. La polimerizzazione radicalica non permette il controllo della tassìa e della regioisomeria, a causa della forte reattività dei radicali. I polimeri a polim radicalica sono: LDPE, PS, PMMA, PTFE, PVC.
Polimerizzazione ionica: il centro attivo è costituito da un catione o anione. Cariche + e - della catena sono sempre affiancate dai propri controioni nella soluzione. Cationica: inizio: I = H+A- quando avvicina a un CH2=CH2 H+ va sulla catena e A- vicino alla carica +: CH3-CH2+ A-, la presenza di H+ sposta i doppietti elettronici del doppio legame: H+ C::C H:C..C+ . Se C+ e A- sono lontani in soluzione polare la reazione procede veloce e incontrollata; propagazione: C+ avvicina un CH3=CH3 e si lega al C spostando di volta in volta la carica lungo la catena. Il termine non può avvenire per acooppiamento ++, il trasferimento di centro attivo fra catene è facile, e quindi ottengo catene corte. Ho controllo sulla regioisomeria dato che + si attacca sempre al C meno ingombro. Anionica: I = CH3- Li+ con CH2=CH2 CH3-CH2-CH2- LI+. E' una reazione completa, tanti I metto tante catene ottengo, perché non c'è trasferimento di centro attivo fra catene. PM è simile per tutte le catene (indice P≈1), dipende da [M] e inversamente da nI, e le catene rimangono sempre attive. Con questa polimerizzazione posso ottenere copolimeri a blocchi, facendo reagire tutto un monomero, poi un altro.
Polimerizzazione Ziegler-Natta: uso catalizzatori ZN: Alluminio alchile AlR3 + Etile -CH2-CH3 + composto di un metallo di transizione (Ti,Zr,V,Mo.) AlEt3 + Ti(OC4H9)4 catalizzatore: *Metallo-CH2-CH3 (ambiente di reazione: H2O, alcoli, acidi, con molta attenzione agli H attivi data la reattività dei catalizzatori). La catalisi può essere omogenea se catalizzatori sono solubili, o eterogenea, con catal. in sospensione. I composti per la reazione sono l'etilene, il propilene, 1-butene, 1,3 butadiene, cicloalcheni. La polimerizzazione è sempre di tipo sequenziale. Il monomero M si inserisce tra il metallo e il resto della catena. Meccanismo di reazione: quando CH2=CH2 si avvicina al cat un C è attratto dal metallo, l'altro dal C legato al metallo, così il legame tra Met-C si rompe e anche il doppio legame C=C, a dare Met-C-C-C. I catalizzatori hanno una geometria che permette il controllo della stereoregolarità, per dare polimeri semicristallini. C'è polidespersità, ma molta regolarità e elevata densità (HDPE).Con ZN ho solo attacco testa-coda dato impossibilità di attacco dove c'è ingombro sterico, e controllo anche la tassìa.
Polimerizzazione a stadi: 1) monomero AA + BB dimero AB reazione di equilibrio (rimangono AA e BB non reagiti); 2) ho varie combinazioni: AB+AA ABA, AB+BB BAB, AB+AB ABAB, AA+BB AB. Per gli stadi successivi ho sempre più combinazioni, e alla fine otterrò una miscela di polimeri di varia lunghezza, con PM basso, e allora devo spingere la conversione verso il 100%. Ci possono essere ramificazioni se A o B presentano più funzionalità, per aumentare la resa devo avere r = rapporto n moli AA / n moli BB ≈ 1, perché il grado di polimerizzazione x = 1+r/1+r-2pr con p=conversione=% di monomeri reagiti. Basta variar di poco p o r per variare di molto x. Esempi: poliammidi, poliesteri, policarbonati, poliuretani.
Poliammidi: reagente base diammina NH2-NH2, reagisce con di acidi HO-C(=O)-R-C(=O)-OH con sottoprodotto H2O, oppure con cloruro acilico Cl-C(=O)-R-C(=O)-Cl a dare HCl, più reattivo del di acido, quindi conversione p maggiore, anche se Hcl è + inquinante. Oppure reagisce con amminoacido: NH2-R-C(=O)-OH + H2N-R-C(=O)-OH via H2O + poliammide; di solito non si fanno reagire amminoacidi aperti ma lattoni, ovvero privo l' amminoacido di H2O e lo chiudo ad anello, cosicché nella reazione H2O funge da catalizzatore per aprire i lattoni e farli reagire. Un esempio di poliammide alifatica è il nylon m,n, in cui m si riferisce al num di C tra i due C(=O) compresi, e n al num di C tra i due N. Più m e n crescono, più rari sono i ponti ad H, più il polimero è vetroso. Le poliammidi sono semicristalline, dati ponti ad H, Tm è circa 600°, sono inerti, e assorbono H2O con fenomeno di autolubrificazione. Un esempio di poliammide aromatica è il Kevlar, in cui R della diammina e del di acido è un fenolo, o il nomex, in cui il fenolo è collegato in meta: hanno forte anisotropia: lungo le fibre alto modulo.
Poliesteri: gruppo estereo -C(=O)-O-. in genera a partire da un diacido HO-C(=O)-R-C(=O)-OH e un di alcol HO-R-OH -C(=O)-R-C(=O)-O-R-O- + H2O. E' una reazione di equilibrio, a T alta e vuoto, poiché dialcolo è volatile e infiammabile, quindi è meglio partire prima con una transesterificazione: 1 diacido e 2 dialcoli a dare HO-R-O-C(=O)-R-C(=O)-R-OH. N.B.: R del di acido è sempre aromatico. Esempi: Polietilentereftalato (PET) parte da acido tereftalico R=fenolo e etilenglicolo R=CH2CH2. Il legame estereo crea legami tra catene di tipo bipolo/bipolo.
Policarbonati: gruppo funzionale -O-C(=O)-O-, a partire da bisfenolo A HO-fen-(CH3-)C(-CH3)-fen-OH + Fosgene Cl-C(=O)-Cl con sottoprodotto HCl.
Poliuretani: gruppo funzionale -NH-C(=O)-O-, è una reazione che fa eccezione perché a stadi ma senza sottoprodotti. Si parte da diisocianato O=C=N-R-N=C=O e dialcol HO-R-OH. Gli isocianati aromatici sono più reattivi degli alifatici, il glicolo ha legami flessibili, così alternando catene flessibili a fenoli rigidi e ponti H ottengo proprietà volute. Applicazioni: assorbimento gas, quindi schiume, espansi.
|
Privacy |
Articolo informazione
Commentare questo articolo:Non sei registratoDevi essere registrato per commentare ISCRIVITI |
Copiare il codice nella pagina web del tuo sito. |
Copyright InfTub.com 2024